US Pharm. 2019;44(8)(Specialty&Oncology suppl):8-10.
ABSTRACT: Transthyretin (TTR) amyloidosis (ATTR) is a progressive, multiorgan, multigenotypic disease caused by TTR amyloid fibril deposition in various tissues. Common clinical manifestations include peripheral and autonomic neuropathy and cardiomyopathy, with substantial morbidity and a poor prognosis. Treatment selection is based on the organs involved; therapies have been limited and frequently have not prevented disease progression. However, in the past year, the FDA has approved several novel treatment options with unique mechanisms of action. Optimal management of ATTR is challenging and requires a multidisciplinary approach. Pharmacists can play a key role in the selection and management of appropriate ATTR treatment.
The amyloidoses are a heterogeneous group of systemic diseases caused by extracellular tissue accumulation of insoluble fibrils that are composed of subunits of misfolded proteins. Amyloid precursor proteins are typically circulating proteins that undergo conformational changes resulting in the formation of beta-pleated sheets. Over time, amyloid deposits can cause cellular death, disruption of normal tissue architecture and function, and eventually death.1 Classification of systemic amyloidoses is based on the misfolded precursor proteins, and at least 30 different precursors have been identified.2 The clinical course, affected organ systems, prognosis, and treatment considerations are determined by the type of precursor protein involved. The most common kinds of acquired amyloidosis are light-chain amyloidosis (caused by deposition of immunoglobulin fragments) and amyloid A amyloidosis (involving acute-phase reactant protein).2
Another clinically important form of amyloidosis is transthyretin (TTR) amyloidosis (ATTR). ATTR is a debilitating, progressive, and eventually fatal multigenotypic disease that is characterized by TTR amyloid fibril deposition in various tissues and affects multiple organs. TTR is intrinsically amyloidogenic, and ATTR can develop through hereditary TTR mutation (hATTR) or secondary to age-related protein misfolding (wild-type ATTR [wtATTR]).3 ATTR can be challenging to diagnose because of its heterogenicity and nonspecific symptoms. Common clinical manifestations include peripheral and autonomic neuropathy and cardiomyopathy, with substantial morbidity and poor prognosis. Optimal management of ATTR is challenging and requires a multidisciplinary approach. Historically, therapeutic options for ATTR have been limited and frequently have not prevented disease progression; however, in the last year, the FDA has approved several first-in-class medications, expanding treatment options for patients. Pharmacists can play a key role in treatment selection, patient and provider education, and supportive-care management.
PATHOPHYSIOLOGY AND CLINICAL MANIFESTATIONS
Formerly known as prealbumin, TTR is a plasma protein responsible for the transport of retinol (vitamin A) and a small fraction (15%) of thyroxine.3 The liver is a major producer of TTR, but small amounts are synthesized by the retinal pigment epithelium and choroid plexus.3 Normally, TTR is a homotetrameric protein; however, because of aging or in the presence of mutation, tetramers can dissociate into dimers and monomers that misfold and form amyloid fibril aggregates.4 ATTR may be fully derived from wtTTR in honhereditary disease (previously called senile amyloidosis) or from a mixture of mutated and wtTTR in hereditary disease.3,4 In both types, the deposition of TTR-derived amyloid in various tissues leads to multisystemic disease. Although multiple organ systems may be affected, mainly the peripheral and autonomic nervous systems and the heart are involved.4,5
In general, wtATTR primarily affects the heart, manifests after an individual’s sixth decade, and is predominant in men.6 Up to 10% of heart-failure (HF) cases in the elderly population are due to wtATTR.7 hATTR, an autosomal-dominant disorder, has considerable variation in penetrance, prevalence, organ-system involvement, and patient outcome depending on the TTR mutation.4 More than 130 mutations in the TTR gene can cause an increase in TTR instability or amyloidogenicity, leading to hATTR.5 Val30Met, the most prevalent TTR mutation worldwide, is typically associated with early-onset neuropathy.8 The Val122Ile mutation is found primarily in African American individuals, and cardiomyopathy is the predominant feature.9 Despite described genotype-phenotype correlations, many hATTR patients present with a mixed clinical phenotype involving neurologic, cardiac, and other system impairment.8 The exact prevalence of hATTR is unknown; it has been reported to affect 50,000 individuals worldwide, but that figure is likely underestimated.4,5
Because of variability in affected organ systems, ATTR amyloidosis can have a heterogeneous clinical presentation manifesting as sensorimotor or autonomic neuropathy, nonspecific cardiac symptoms, or—more rarely—renal or ocular impairment.4,5,8 Sensory neuropathy is progressive, initially affecting the lower limbs and eventually reaching the upper limbs.5 Symptoms include neuropathic pain, numbness, and impaired thermal sensitivity. Motor impairment typically occurs in the lower limbs first, starting with difficulty walking and loss of balance and eventually rendering the patient wheelchair- or bed-bound.5 Patients may develop autonomic neuropathies such as sweating abnormalities, erectile dysfunction (ED), urinary retention, orthostatic hypotension (OH), and disturbed gastrointestinal (GI) motility.5,8 Amyloid deposits in the heart cause ventricular-wall thickening and diastolic dysfunction, arrhythmias, and ultimately HF.5,8 Early disease is usually minimally symptomatic, with edema, dyspnea, progressive fatigue, elevated jugular venous distention, and other HF symptoms developing with disease progression. Carpal tunnel syndrome may be an early but nonspecific manifestation of hATTR, a circumstance often overlooked by clinicians.9
Owing to its nonspecific and variable presentation, ATTR is often misdiagnosed (e.g., as chronic inflammatory demyelinating polyneuropathy or hypertrophic cardiomyopathy).4 However, early diagnosis and treatment are essential because the symptoms can progress rapidly.
TREATMENT
Symptom Management
Symptom management is a crucial aspect of care for ATTR patients.4,8 In those with polyneuropathy, medications for neuropathic pain (e.g., gabapentin, pregabalin, duloxetine) and control of autonomic dysfunction (e.g., antidiarrheals, GI-motility medications) can significantly improve quality of life (QoL).10 Phosphodiesterase inhibitors should be used with caution in patients with ED owing to the risk of worsening hypotension.8 Fluid balance and reduction of filling pressure are key goals for supportive management of ATTR cardiomyopathy.4,6,8 Although no randomized, controlled trials have been conducted on the management of amyloidosis-induced HF, low-dose loop diuretics with or without aldosterone antagonists are the mainstay of therapy.6,8 Many agents commonly used to manage HF are poorly tolerated or not beneficial in amyloidosis; for example, calcium channel blockers and digoxin may lead to symptom exacerbation because they bind to amyloid fibrils.4,6 Patients with cardiac amyloidosis may be particularly sensitive to beta-blockers, ACE inhibitors, and angiotensin receptor blockers; they may develop profound hypotension, fatigue, or worsening OH even at low doses.6 Amiodarone or a permanent pacemaker may be used to manage arrhythmias. However, studies have not demonstrated a survival benefit with implanted cardiodefibrillators for primary or secondary prevention.6
Organ Transplantation
Orthotopic liver transplantation (OLT), the first disease-modifying treatment, was introduced in the 1990s.8 Because most TTR is produced by the liver, OLT significantly reduces the production of mutant TTR, replacing it with the donor’s wt protein. OLT reduces variant TTR concentrations by up to 98% and significantly increases patient survival.11 It seems to be particularly beneficial for patients with Val30Met mutations and early-stage disease, preventing progressive neuropathy and resulting in excellent survival rates.8,11 Outcomes in patients with other mutations, however, seem to be markedly worse.4,8 Additionally, cardiomyopathy and neuropathy can progress owing to wtTTR complexing with already-existing amyloid deposits.4,8 Therefore, combined liver and heart transplantation may be considered for patients with preexisting ATTR cardiomyopathy.6 Such surgery carries substantial risk, however, and conflicting data regarding patient outcomes have been reported. Some studies have shown outcomes similar to those for general heart-transplant populations, whereas others have reported shorter survival and worse outcomes.6 Because OLT does not act on the production of mutant TTR in the choroid plexus and retinal pigmented epithelium, progression or development of central nervous system (CNS) or ocular manifestations does not occur.8 The utility of OLT is limited by patient eligibility, organ availability, and the risk of adverse events. Because of morbidity from the procedure itself, some patients did not report an improvement in QoL despite slower disease progression.8
Targeted Therapies
TTR Stabilization: Because dissociation of TTR into two dimers is the rate-limiting step in the formation of amyloid fibrils, an alternative approach is to stabilize the circulating TTR protein to prevent its dissociation.4,8 The two available therapies that aim to stabilize TTR are diflunisal and tafamidis.
Diflunisal: Diflunisal is a nonsteroidal anti-inflammatory drug (NSAID) that stabilizes TTR by binding to its thyroxine-binding sites. Although it is not approved for ATTR, diflunisal may be given off-label.4,8 In a randomized, placebo-controlled trial of 130 hATTR patients with polyneuropathy after 2 years of therapy, diflunisal decreased progression of neuropathy and improved QoL compared with placebo.12 Among patients with baseline cardiac involvement, diflunisal treatment did not significantly decrease left ventricular (LV) wall thickness or LV longitudinal strain versus placebo.10 Diflunisal administered at a dosage of 250 mg orally twice daily was well tolerated in this trial, but cases of renal dysfunction and thrombocytopenia have been reported in other studies.10 In general, NSAID therapy is associated with a risk of GI, renal, and cardiac side effects and is contraindicated in patients with renal failure or HF. Therefore, the use of diflunisal may be limited in hATTR patients who have severe ATTR cardiomyopathy. Careful patient selection and surveillance are important until long-term safety data become available.
Tafamidis: This agent (TABLE 1), a small-molecule TTR stabilizer, was recently FDA-approved to treat cardiomyopathy of wtATTR or hATTR in adults. In Europe, it is approved to delay peripheral neurologic impairment in adults with hATTR and stage 1 symptomatic polyneuropathy.4 The drug’s efficacy for hATTR with polyneuropathy was assessed in a placebo-controlled phase II/III trial in patients with Val30Met mutations and stage 1 disease.13 The trial did not meet its coprimary endpoints of <2-point worsening of Neuropathy Impairment Score (NIS)–Lower Limbs and change from baseline in Norfolk Quality of Life–Diabetic Neuropathy (Norfolk QoL-DN) total QoL score (TQoL) in the intent-to-treat population at 18 months (45.3% vs. 29.5%; P = .068).13 However, in an efficacy-evaluable population, significantly more patients achieved treatment response and better preservation of TQoL with tafamidis compared with placebo (60% vs. 38.1%; P = .041 and 0.1 vs. 8.9 points; P = .045, respectively).13 Unfortunately, tafamidis’s effect on decrease in disease progression was less pronounced in patients with more advanced, late-onset disease or mutations other than Val30Met.14 More encouraging results leading to FDA approval were demonstrated in the phase III ATTR-ACT trial in wtATTR and hATTR patients with predominantly cardiac manifestations.14 Compared with placebo, tafamidis significantly reduced all-cause mortality and cardiovascular-related hospitalizations at 30 months (29.5 vs 42.9%; hazard ratio, 0.70; 95% CI, 0.51-0.96 and 0.48/year vs. 0.7/year; relative risk reduction, 0.68; 95% CI, 0.56-0.81, respectively).14 Significant differences were observed in key secondary endpoints such as the 6-minute walk test and QoL.14 Subgroup analysis demonstrated greater benefit in patients with less severe HF, suggesting that initiation of tafamidis at an early disease stage may be most beneficial.14 Based on current data, tafamidis is generally well tolerated, with a safety profile similar to that of placebo.4,13-15
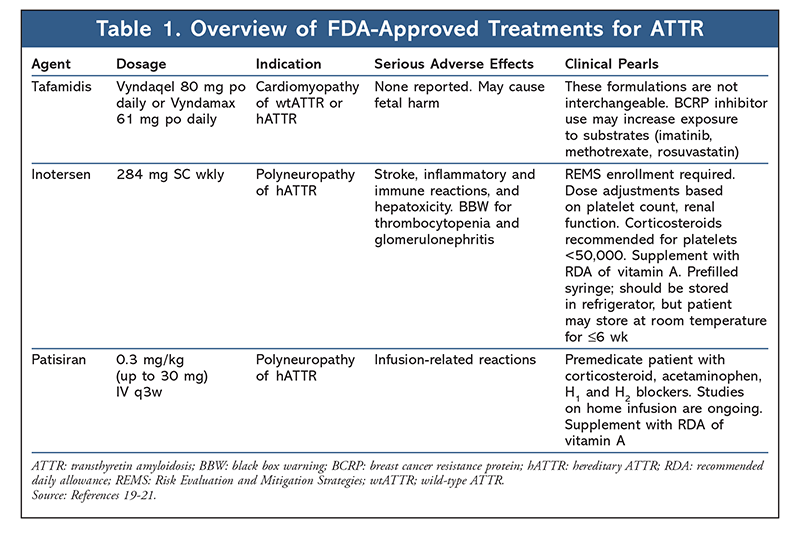
Gene-Modifying Therapy: Laboratory data suggest that the presence of circulating TTR is not required for life; therefore, the knockdown of TTR gene expression is not expected to be harmful to patients.16 In the past year, two gene-silencing treatments have been FDA-approved to treat hATTR polyneuropathy: inotersen (antisense oligonucleotide [ASO] and patisiran (small interfering RNA [siRNA]) (table 1). RNA interference is a natural catalytic mechanism that results in the knockdown of target messenger RNA (mRNA) by siRNA bound to the silencing complex.4,17 Patisiran siRNA targets the 3´ untranslated region of TTR mRNA devoid of any known TTR mutations, thus preventing production of wt and mutated protein.17 Encapsulation with lipid nanoparticles enables siRNA delivery to the site of action (hepatocytes).4 Inotersen, a second-generation ASO, also binds at the 3´ untranslated region of TRR mRNA, resulting in mRNA degradation.18 The two pivotal phase III trials (NEURO-TTR and APOLLO) that led to approval of these agents were similar in design, study population, and outcome measures.17,18 In both trials, patients receiving the active agent had improved clinical manifestations of disease as measured by modified Neuropathy Impairment Score + 7 tests and Norfolk QoL-DN.17,18 Although the safety profile of patisiran was similar to that of placebo, some potential safety concerns were noted in the inotersen trial. That trial found increased risks of thrombocytopenia and glomerulonephritis, necessitating increased monitoring through a restricted-distribution Risk Evaluation and Mitigation Strategies program.19
Agents in Development: Other agents in various stages of development include novel TTR stabilizers designed with higher tetramer-stabilization properties, anti-TTR antibodies, and anti-serum amyloid P agents.4,6 In mouse models, a combination of doxycycline (capable of disrupting TTR fibril formation) and tauroursodeoxycholic acid (can reduce nonfibrillar TTR deposition) has shown synergy in reducing TTR deposits.4 Antiparkinson therapy (tolcapone) has been shown to stabilize TTR tetramers; this could be an attractive option for ATTR with leptomeningeal involvement because tolcapone is known to cross the blood-brain barrier.4 However, data from large randomized trials of these agents are lacking.
CONCLUSION
ATTR is a complex, progressive, and debilitating disease characterized by peripheral and autonomic neuropathy and cardiomyopathy caused by deposition of wtTTR or mutant TTR amyloid fibrils. Current treatments include liver transplantation, TTR stabilizers, and gene-modifying therapies. Symptom management is important for maintaining QoL, and disease-drug interactions must be addressed. Pharmacists are well positioned to provide appropriate symptom and treatment management for patients with ATTR.
REFERENCES
1. Chiti F, Dobson CM. Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu Rev Biochem. 2017;86:27-68.
2. Benson MD, Buxbaum JN, Eisenberg DS, et al. Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid. 2018;25:215-219.
3. Vieira M, Saraiva MJ. Transthyretin: a multifaceted protein. Biomol Concepts. 2014;5:45-54.
4. Hawkins PN, Ando Y, Dispenzeri A, et al. Evolving landscape in the management of transthyretin amyloidosis. Ann Med. 2015;47:625-638.
5. Coelho T, Maurer MS, Suhr OB. THAOS—The Transthyretin Amyloidosis Outcomes Survey: initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr Med Res Opin. 2013;29:63-76.
6. Siddiqi OK, Ruberg FL. Cardiac amyloidosis: an update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med. 2018;28:10-21.
7. González-López E, Gallego-Delgado M, Guzzo-Merello G, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36:2585-2594.
8. Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8:31.
9. Maurer MS, Hanna M, Grogan M, et al. Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol. 2016;68:161-172.
10. Adams D. Recent advances in the treatment of familial amyloid polyneuropathy. Ther Adv Neurol Disord. 2013;6:129-139.
11. Adams D, Samuel D, Goulon-Goeau C, et al. The course and prognostic factors of familial amyloid polyneuropathy after liver transplantation. Brain. 2000;123:1495-1504.
12. Berk JL, Suhr OB, Obici L, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310:2658-2667.
13. Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79:785-792.
14. Lozeron P, Theaudin M, Mincheva Z, et al. Effect on disability and safety of Tafamidis in late onset of Met30 transthyretin familial amyloid polyneuropathy. Eur J Neurol. 2013;20:1539-1545.
15. Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379:1007-1016.
16. Episkopou V, Maeda S, Nishiguchi S, et al. Disruption of the transthyretin gene results in mice with depressed levels of plasma retinal and thyroid hormone. Proc Natl Acad Sci U S A. 1993;90:2375-2379.
17. Adams D, Gonzalez-Duarte A, O’Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379:11-21.
18. Benson MD, Waddington-Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379:22-31.
19. Tegsedi (inotersen) package insert. Carlsbad, CA: Ionis Pharmaceuticals, Inc; October 2018.
20. Onpattro (patisiran) package insert. Cambridge, MA: Alnylam Pharmaceuticals, Inc; August 2018.
21. Vyndaqel (tafamidis meglumine) and Vyndamax (tafamidis) package insert. New York, NY: Pfizer Inc; May 2019.
To comment on this article, contact rdavidson@uspharmacist.com.



