US Pharm. 2016;41(6):HS-11-HS-13.
Sjögrin syndrome, pronounced SHOW-grin), or SS, is an autoimmune disease characterized by parotid gland enlargement and dryness of the mouth and eyes. The disease was first identified by a Swedish physician, Henrik Sjögren, in 1933. It is a systemic, chronic inflammatory disorder characterized by lymphocytic infiltrates in exocrine organs. Inflammation of the lacrimal glands leads to decreased tear production, while inflammation of the salivary glands leads to decreased saliva production and dry lips. Diagnosis of SS is difficult and may take several years through many laboratory tests and diagnostic procedures.1
All autoimmune diseases are caused by the abnormal production of extra antibodies in the blood that are directed against various tissues of the body. The misdirected immune system in autoimmunity tends to lead to inflammation of tissues.
SS is one of the most prevalent autoimmune disorders, striking as many as 4 million Americans. Nine out of 10 patients are women, and the average age of diagnosis is around 40 years, although the syndrome can occur in all age groups and in both sexes. Early diagnosis and proper treatment may prevent serious complications and greatly improve quality of life for individuals living with this autoimmune disease. The inflammation of parotid glands in mumps is due to a viral infection and will not be discussed here. In this article, we will briefly review the clinical presentations of SS, with an emphasis on its diagnosis and symptomatic treatment.2
Clinical Presentations
SS can affect all of the moisture-producing glands and tissues in the body and cause a host of symptoms. These symptoms vary from person to person and may include a dry or burning sensation in the eyes, dry mouth, difficulty talking or swallowing, a cracked tongue, dry or burning throat, dry and peeling lips, a change in taste or smell, increased dental decay, joint pain, vaginal dryness, digestive problems, and dry nose. About 50% of patients with SS have skin issues, such as dryness and/or urticaria.3
SS may cause dysfunction of other organs, affecting the kidneys, gastrointestinal system, blood vessels, lungs, liver, pancreas, and nervous system. Patients also have a higher risk of developing lymphoma. About 50% of the time SS occurs alone (primary), and in the other 50% it occurs in the presence of another connective tissue disease (secondary) such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), or scleroderma. SS is systemic and affects the entire body; however, symptoms may remain low-intensity, worsen, or go into remission. While some patients experience mild discomfort, others have severe symptoms that greatly impair their daily activities. The symptoms of SS may mimic those of medical conditions such as SLE, RA, fibromyalgia, chronic fatigue syndrome, and multiple sclerosis.4
Sjögren Antibodies
SS-A/Ro and SS-B/La are two ribonucleoproteins with small amounts of RNA circulating in the cytoplasm of cells found throughout the body. The autoantibodies against SS-A/Ro and SS-B/La are strongly associated with SS and SLE.5 The presence of these autoantibodies helps aid in the diagnosis of both conditions.
The SS-A/Ro autoantibodies are detected in 40% to 90% of patients with SS and in 20% to 60% of SLE patients. The SS-B/La autoantibodies rarely occur in the absence of SS-A/Ro antibodies, and their presence is more associated with SS. They are found in approximately 60% of patients with SS and 5% to 35% of those with SLE.
Although the presence of SS-A/Ro and SS-B/La antibodies is used to support the diagnosis of SS, it must be interpreted in the proper clinical context because these antibodies are also found in some patients with SLE and other connective-tissue diseases.4
Most investigators believe that a positive result for SS-A/Ro autoantibodies is consistent with connective tissue disease, including SS, SLE, and RA.5
Diagnosis and Classifications
In many cases, SS is undiagnosed or misdiagnosed, but if diagnosis is successful, early interventions are very important and can prevent serious complications and greatly improve a patient’s quality of life.6
The American-European criteria for the classification of SS were proposed in 2002 and are the most commonly used criteria for diagnosis. However, in 2012, a new set of classification criteria was developed by the Sjögren’s International Collaborative Clinical Alliance (SICCA) investigators and accepted as a provisional criteria set by the American College of Rheumatology (ACR).3
Primary SS occurs in the absence of another underlying rheumatic disorder, whereas secondary SS is associated with another underlying rheumatic disease, such as SLE, RA, or scleroderma. Given the overlap of SS with many other rheumatic disorders, it is sometimes difficult to determine whether a clinical manifestation is solely a consequence of SS or is due to one of its overlapping disorders.3
The average time from the onset of symptoms to diagnosis is >4 years. Because all symptoms are not always present at the same time and because SS can involve several body systems, physicians sometimes treat each symptom individually and do not recognize that a systemic disease is present.7
According to the American-European classification system, diagnosis of primary SS requires at least four of the criteria listed in SIDEBAR 1; in addition, either criterion 5 or criterion 6 must be included. Secondary SS is diagnosed when, in the presence of a connective-tissue disease, symptoms of oral or ocular dryness exist in addition to criterion 3, 4, or 5.
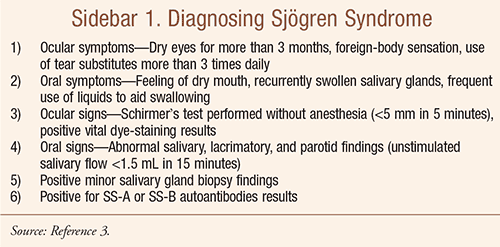
Application of these criteria has yielded a sensitivity of 97.2% and a specificity of 48.6% for the diagnosis of primary SS and a sensitivity of 97.2% and a specificity of 64.7% for diagnosis of secondary SS.
The ACR classification criteria were developed by SICCA investigators in an effort to improve specificity of criteria used for entry into clinical trials, especially in light of the emergence of biologic agents as potential treatments for SS and its associated comorbidities. This high specificity makes the ACR criteria more suitable for application in situations in which misclassification may present a health risk.3
According to the ACR criteria, the diagnosis of SS requires at least two of the following three findings: Positive serum autoantibodies to SS-A and/or SS-B or positive rheumatoid factor and antinuclear antibody titer of at least 1:320; ocular staining score of at least 3; and presence of focal lymphocytic sialadenitis with a focus score of at least 1 focus/4 mm2 in labial salivary gland biopsy samples.
The ACR criteria are based entirely on a combination of objective tests that assess the three main components of SS (serologic, ocular, and salivary) and do not include criteria based on subjective symptoms of ocular and oral dryness.
Application of these criteria has yielded a sensitivity of 93% and a specificity of 95% for the diagnosis of SS. These criteria do not distinguish between primary and secondary forms of the disease.8
Treatment and Management
Treatment for SS is largely based on symptoms (e.g., lotion for dry skin, artificial tears for dry eyes).9,10 Rituximab has shown promise in the treatment of extraglandular manifestations such as vasculitis, cryoglobulinemia, and peripheral neuropathy. This drug is used to restore normal functioning of the immune system in a dosage of 1 g every 2 weeks on Days 1 and 15. Patients must be monitored carefully for the potential development of lymphoma, as the risk for this disease is significantly higher than in the general population.
Artificial saliva products often provide relief from dry mouth (e.g., Mouthkote and Salivert). Toothpastes and mouth rinses have been developed specifically for dry mouth such as Biotene and Orajel. In cases where replacing moisture does not relieve dryness, physicians may prescribe a medication to stimulate the patient’s own salivary glands to produce more saliva. Two such drugs, pilocarpine and cevimeline, are approved for dry mouth associated with SS.
Artificial tears such as Nature’s Tears and Lacril can relieve dry eyes and help prevent damage caused by dryness. If a patient cannot achieve relief from these products, the immunosuppressive drug cyclosporine in the form of an eye drop (Restasis) can be prescribed and applied every 12 hours. A cellulose pellet (Lacrisert) may also be placed in the lower eyelid where it dissolves, adding moisture when artificial tears are used.
The two drugs approved for dry mouth, pilocarpine and cevimeline, may also be useful for dry eyes. While they have not been approved for dry eyes, some physicians prescribe them off-label for that purpose. In severe cases, physicians may consider an outpatient procedure called punctal occlusion, which involves cauterizing the small openings at the inner corners of the eyelids where tears drain from the eyes to close them, keeping the eye’s natural tears on the surface longer.
In some patients, a lack of saliva, which neutralizes stomach acids, can cause heartburn. In these cases, sucralfate, an ulcer medication, can coat and protect the esophagus and stomach. H2-receptor antagonists such as famotidine reduce the production of gastric acid as well. Proton pump inhibitors such as pantoprazole also decrease the amount of acid in the stomach and intestines.
Dry nasal passages can make sinus infections last longer and lead to complications such as pneumonia and bronchitis. Saline nasal sprays and decongestants can be used.
Treatment for dry skin typically consists of topical creams and lotions. If the corners of the lips crack, the cause could be a yeast infection. Topical antifungals such as econazole and miconazole may be needed to clear the infection.
Vaginal lubricants and moisturizers can restore moisture and make intercourse easier. There are many different vaginal lubricants available OTC as gels, sprays, and inserts.
Treatment for joint inflammation typically consists of nonsteroidal anti-inflammatory drugs such as ibuprofen. For more severe inflammation, a corticosteroid medication such as prednisone, which mimics natural substances that control immune response, may be used. Disease-modifying antirheumatic drugs (DMARDs) such as hydroxychloroquine and methotrexate can inhibit the body’s immune response and be used in severe cases.
SS can cause scarring of the lungs, which cannot be reversed, but medical treatment may help prevent it from progressing. Treatment may consist of a combination of prescription medications and medical procedures. Corticosteroids, DMARDs such as azathioprine or cyclophosphamide, the mucus-thinning medication acetylcysteine, antifibrotic (antiscarring) medications such as bosentan and pirfenidone, and oxygen therapy may make breathing less difficult. Pulmonary rehabilitation will also improve breathing to become more efficient.
The body-wide inflammation of SS can affect the nervous system as well. Corticosteroids to treat nerve inflammation and analgesics to relieve pain may be used.
Finally, SS can cause inflammation in the kidneys and their structures. When necessary, treatment may consist of alkaline agents to maintain the balance of blood chemicals normally handled by the kidneys.
The newly established Sjögren’s Center at Johns Hopkins University has more guidelines and information on this autoimmune disease, and patients can be referred to this center for the most up-to-date information about their condition.11
Monitoring
Most patients with SS can be monitored at follow-up visits every 3 months and, if they are stable, up to every 6 months for the progress of all clinical symptoms. Patients with active problems and associated illness must be seen monthly. Patients should be encouraged to avoid factors that cause dryness symptoms such as smoking or exposure to low-humidity environments. All patients with SS should be monitored by an ophthalmologist, dentist, and rheumatologist.
REFERENCES
1. Jonsson R, Vogelsang P, Volchenkov R, et al. The complexity of Sjogren’s syndrome: novel aspects on pathogenesis. Immunol Lett. 2011;141(1):1-9.
2. Mavragant CP, Moutsopoulos NM, Mountsopoulos HM. The management of SS. Nat Clin Pract Rheumatol. 2006;2:252-261.
3. Baer AN. Diagnosis and classification of Sjogren’s syndrome. In: Post TW, ed. UpToDate. Waltham, MA: UpToDate; 2015. www.uptodate.com. Accessed April 1, 2016.
4. Fox R. Clinical manifestations of Sjögren’s syndrome: exocrine gland disease. In: Post TW, ed. UpToDate. Waltham, MA: UpToDate; 2015. www.uptodate.com. Accessed April 1, 2016.
5. Bloch DB. The anti-Ro/SSA and anti-La/SSB antigen-antibody systems. In: Post TW, ed. UpToDate. Waltham, MA: UpToDate; 2015. www.uptodate.com. Accessed March 27, 2016.
6. Venables PJ. Sjogren’s syndrome. Best Pract Res Clin Rheumatol. 2004;18:313-329.
7. Lazarus MN, Isenberg DA. Development of additional autoimmune disease in a population of patients with primary SS. Ann Rheum Dis. 2005;64:1062-1064.
8. Ramos-Casals M, Brito-Zeron P, Font J. The overlap of SS with other systemic autoimmune diseases. Semin Arthritis Rheum. 2007;36:246-255.
9. Akpek EK, Lindsley KB, Advanthaya RS, et al. Treatment of Sjogren’s syndrome. Opthamology. 2011;118(7):1242-1252.
10. Arthritis foundation. www.arthritis.org/about-arthritis/types/sjogrens-syndrome. Accessed March 27, 2016.
11. Johns Hopkins Sjogren Center, 2015. www.hopkinssjogrens.org. Accessed April 1, 2016.



