US Pharm. 2023;48(7):33-38.
ABSTRACT: Non–small cell lung cancer (NSCLC), the predominant form of lung cancer, carries a poor prognosis. Metastatic NSCLC is treated with molecularly targeted therapy and/or immunotherapy to combat complex genetic mutation and immunosuppressive events that drive malignancy. However, these therapies have toxicities as well as poor clinical outcomes. Investigational treatments such as combination therapy target epigenetic dysregulation of cell growth and immune modulation. Preclinical data suggest that trametinib-entinostat combination therapy shows promise in reducing tumor burden in NSCLC associated with co-mutations in an oncogenic driver and tumor-suppressor gene. Oncology pharmacists can identify drug-related problems associated with conventional and emerging therapies for NSCLC and provide education about side-effect profiles, monitoring parameters, and potential drug interactions.
Non–small cell lung cancer (NSCLC) is the most prevalent type of lung cancer, accounting for approximately 85% of cases.1 The histologic subtypes of NSCLC are adenocarcinoma, squamous cell carcinoma, and large cell carcinoma.2 Lung cancer is the leading cause of cancer-related death in both men and women worldwide; only 21.7% of lung cancer patients survive 5 years or more after diagnosis.3,4 Therefore, early diagnosis, staging, and delivery of precision therapy are essential to reduce mortality.1,5,6
The prevalence of NSCLC increased between 2010 and 2016, with more cases occurring in women than in men.5 NSCLC has a substantially higher prevalence in individuals aged 65 years and older; alarmingly, however, the number of cases in persons younger than age 65 years has risen.5 Although a decrease in the smoking rate reduced NSCLC incidence in both sexes across all ages between 2010 and 2017, the incidence remains higher in men and in older patients.2,5 Increased screening and detection have resulted in a concomitant rise in the incidence of stage I NSCLC as well as decline in the incidence of stage IV NSCLC.5 The survival rate for NSCLC decreases with each year following diagnosis and worsens with advanced stage at time of diagnosis.4
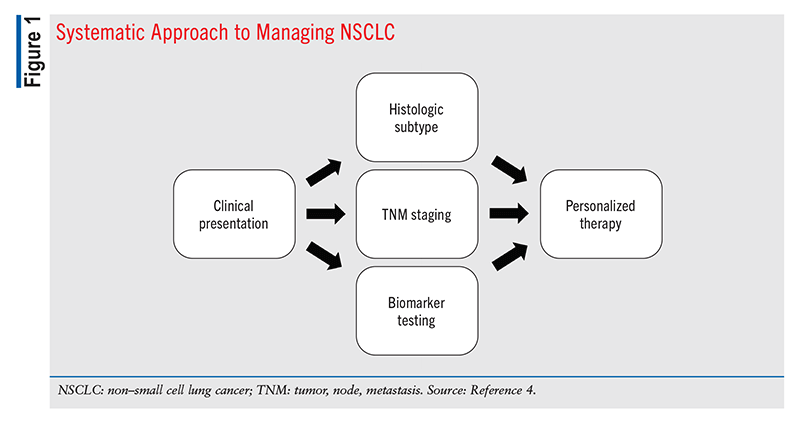
Metastatic lung cancer patients who are eligible for targeted therapies or immunotherapies (vs. conventional radiotherapy and chemotherapy) currently have a 5-year survival rate ranging from 15% to 50%. In metastatic NSCLC, early determination of histologic subtype, identification of tumor-specific and immune biomarkers, and appropriate selection of personalized therapy are critical to improve patients’ overall survival (OS) (FIGURE 1).3,4 Deficiency in the tumor suppressor liver kinase B1 (LKB1) accelerates tumor growth in NSCLC malignancies with oncogenic driver mutations.7 Kinase overactivity promotes tumor growth, and the use of kinase inhibitors (e.g., tyrosine kinase inhibitors [TKis] for receptor tyrosine kinase [RTK]) has been a mainstay of NSCLC treatment. Another major therapeutic approach has been to target histone deacetylases (HDACs) in order to improve the immune system’s ability to recognize and attack tumor cells and inhibit cell proliferation.8
This article will review the risk factors for developing NSCLC and how this type of cancer is staged and diagnosed. It will also discuss the utility of conventional treatments for NSCLC as well as emerging combination therapies, including the combination of trametinib (a mitogen-activated protein kinase [MEK] inhibitor [MEKi]) and entinostat (a class I HDAC inhibitor [HDACi]). Promising preclinical data indicate that combination treatment with trametinib and entinostat effectively combats the LKB1 mutation in NSCLC and could potentially improve OS. As the medication experts on the healthcare team, oncology pharmacists stand equipped to identify drug-related problems associated with conventional NSCLC treatments as well as emerging combination therapies for NSCLC, such as trametinib-entinostat, and to educate patients and healthcare providers about side-effect profiles, monitoring parameters, and potential drug interactions.
Risk Factors, Diagnosis, and Staging
Both modifiable risk factors (e.g., exposure to carcinogens) and nonmodifiable risk factors (e.g., genetic susceptibility) increase the chance of developing NSCLC (TABLE 1). Cigarette smoking is the leading risk factor, with smoking history (number of cigarettes, tar and nicotine content, length of time) being directly proportional to risk.2 Pulmonary fibrosis and HIV also confer an increased risk of developing NSCLC.9,10 The presence of risk factors, combined with clinical manifestation of respiratory symptoms, warrants lung cancer screening.

Diagnosis and staging entail determining the tumor location, tissue of origin, and extent of spread by performing a physical examination, tissue biopsy, clinical laboratory testing (CBC, electrolytes, creatinine, blood urea nitrogen), and multiple imaging procedures (chest radiography, computed tomography, positron emission tomography, magnetic resonance imaging).2,6 The TNM staging system is commonly used to ascertain tumor size (T), whether cancer has spread to lymph nodes (N), and extent of metastasis to distant sites (M).11 Stages range from 0 to IV; stage 0 signifies in situ disease and carries the best prognosis, whereas stage IV indicates metastasis and carries the worst prognosis.11 Most NSCLC patients present with locally advanced or metastatic disease at the time of diagnosis.1 Along with diagnosis and staging, the histologic subtype and genetic mutations specific to the individual tumor are identified. This information is used collectively to direct personalized treatment plans. Precision therapies based on specific genetic mutations within tumor cells reduce symptoms, tumor-mutation burden, and OS, and they improve the patient’s quality of life.4
Etiology
Genetic: DNA mutations are a common factor in lung cancer. Common oncogenic drivers involve mutations in (in order of prevalence) Kirsten rat sarcoma virus (KRAS); epidermal growth factor receptor (EGFR); anaplastic lymphoma kinase (ALK); ROS proto-oncogene 1 (ROS1); RET proto-oncogene; proto-oncogene B-Raf (BRAF); human epidermal growth factor receptor 2; neurotrophic tyrosine kinase 1, 2, and 3; mesenchymal-epithelial transition factor; and fibroblast growth factor receptor (FGFR) 1 (TABLE 1).12
KRAS mutations occur in 20% to 40% of lung adenocarcinomas, and some patients exhibit co-occurring oncogenic targets.13-15 Genetic mutations of the tumor suppressor LKB1 occur in 20% to 30% of NSCLC cases. LKB1-mutant tumors are associated with poor prognosis, as they are highly aggressive and resistant to chemotherapy.16-18 LKB1 is a serine/threonine kinase that activates adenosine monophosphate–activated protein kinase (AMPK) by phosphorylating Thr172 within the AMPK-alpha activation loop.19 AMPK serves as a master nutrient sensor that regulates cellular metabolism. When energy availability (i.e., adenosine triphosphate) is low, or during hypoxic stress, AMPK becomes activated. In response to metabolic stress, AMPK stimulates energy-generating catabolic processes such as glucose uptake and lipid oxidation while inhibiting energy-consuming anabolic processes in order to restore cellular metabolic homeostasis. By inhibiting cell growth and proliferation and promoting energy conservation, AMPK signaling is antitumorigenic.20 When LKB1 is mutated, AMPK signaling is impaired, resulting in metabolic reprogramming (e.g., hyperglycemia, increased fatty acid synthesis and dependence on glutamine, decreased oxidation) and excessive cell growth and division.7,21,22 LKB1-mutant tumors are also more resistant to immunotherapy, and affected individuals are more likely to experience localized recurrence and mortality.22 Approximately one-half of LKB1 mutations occur concurrently with KRAS mutations, rendering this type of double-mutant tumor especially metastatic and difficult to treat.22,23
Epigenetic: Aberrations in tumor-suppressor genes or oncogenes associated with NSCLC are not always due to genetic mutations; they are sometimes attributable to alterations in gene transcription arising from epigenetic events (TABLE 1). Unlike genetic mutations, epigenetic modifications (e.g., acetylation, methylation, phosphorylation) are reversible because they do not involve alterations in the DNA sequence and are enzymatically regulated posttranslationally. HDACs epigenetically regulate gene expression by removing acetyl groups from histones, highly basic proteins that associate with DNA in the nucleus and help condense it into chromatin. Deacetylation inhibits gene transcription, whereas acetylation activates gene transcription.24 Modulation of gene transcription, in turn, regulates the expression levels and functions of various target genes and proteins that either promote or suppress tumor growth. Class I HDACs include HDAC1, 2, 3, and 8. Located in the cell nucleus, they play key roles in regulating the cell cycle; cell differentiation, proliferation, and apoptosis; metastasis; and angiogenesis. HDAC1 is critical for immunomodulatory processes, whereas HDAC3 regulates cell growth. Increased HDAC1 gene expression is associated with lung cancer progression in preclinical studies, a large percentage of NSCLC tumors display elevated HDAC3 expression, and histone hypoacetylation is linked to a more aggressive adenocarcinoma phenotype in the lung.24-26
Management
Targeted Therapy Versus Immunotherapy: NSCLC management is based on tumor staging, histologic subtype, and genetic mutation. Stage I and II NSCLC cases are primarily treated surgically.27 Metastatic NSCLC occurs with or without an oncogenic driver, and pharmacologic treatment regimens are tailored accordingly.28 Frequently, genetic mutations involve dysregulated RTKs (e.g., RAS, BRAF, and MEK), which promote uncontrolled cell growth and proliferation through hyperactivation of the mitogen-activated protein kinase cell signaling pathway.29,30 Targeted therapies use small-molecule drugs that inhibit the molecular pathways responsible for tumor growth and maintenance. These agents are therefore reserved for patients who test positive for EGFR-activating mutations or ALK/ROS1 translocations.31 TKis are a mainstay of targeted treatment for EGFR-mutant NSCLC. However, the targeting of KRAS-mutant NSCLC (directly with farnesyl transferase inhibitors, inhibitors of downstream MEK, and synthetic lethality) has largely failed.15 Pharmacologic therapies that are effective against LKB1-mutant tumors include biguanides (which compromise mitochondria and reduce systemic glucose availability), glutaminase inhibitors (which inhibit tricarboxylic acid cycle progression), and nutrient deprivation.18 Inhibition of proteins downstream from LKB1—such as mammalian target of rapamycin, MEK, and extracellular signal-regulated kinase (ERK)—has been another preclinical approach.18 Interestingly, radiosensitization (the phenomenon whereby tumor cells become more sensitive to radiotherapy) by the MEK inhibitor trametinib induces senescence only in KRAS-LKB1–mutated cells exhibiting normal tumor-suppressor protein p53 expression.32
Alternatively, immunotherapy employs the use of monoclonal antibodies directed against immune checkpoint molecules, such as receptor programmed cell death 1 (PD-1) or its ligand (PD-L1).31,33 PD-L1 facilitates tumor immune evasion by suppressing immunosurveillance by T lymphocytes. Immune checkpoint inhibitors (ICIs) block the interaction between PD-1 and PD-L1, thereby enabling the immune system to detect and eliminate cancer cells; ICIs are therefore used in PD-L1–positive patients.31 When an oncogenic driver is absent, ICI monotherapy or combination therapy with platinum-based chemotherapy is standard.34,35 Interestingly, ICIs improve OS compared with standard chemotherapy in KRAS-mutant, but not wild-type, NSCLC.15,36
MEKis: MEK1 and MEK2 are upstream regulators of the ERK signaling pathway that relay signals promoting cellular proliferation. Trametinib (a reversible inhibitor of MEK1 and MEK2) and other MEKis block MEK-dependent signaling to promote cell death, arrest cell growth, and inhibit tumor growth. Unfortunately, MEKi monotherapy for NSCLC has been unsuccessful in clinical trials to date compared with chemotherapy alone, resulting in poor clinical outcomes and increased toxicities.37-40 Toxicities associated with MEKi therapy include dermatitis acneiform, rash, nausea, vomiting, diarrhea, fatigue, and hypertension.38-40 Trametinib may also be administered in combination with chemotherapy, ICIs, EGFR-TKis, and other anticancer agents, with variable results depending upon the genetic mutation.37 For instance, the combination of the BRAF inhibitor dabrafenib and trametinib is first-line treatment for advanced BRAF-mutant NSCLC, but OS remains poor.41-43
One major challenge to MEKi therapy (along with other drugs that target RAS/RAF/MEK/ERK signaling) is the development of drug resistance mutations.44,45 In KRAS-mutant NSCLC, trametinib resistance is attributed to compensatory reactivation of FGF21, the expression of which is upregulated in lung cancer tissues and cell lines. Overactive FGF21 promotes growth, migration, and tolerance to oxidative stress, thereby creating a favorable tumor microenvironment (TME).46 Combination treatment with trametinib and the TKi ponatinib, which blocks signaling at the FGFR, is associated with cardiovascular and bleeding toxicities in patients with KRAS-mutant NSCLC.43 Diarrhea and rash also contribute to difficulty tolerating this combination treatment.43
HDACis: HDACis induce histone hyperacetylation and transcriptional activation of genes that suppress cell proliferation. Several HDACi classes, distinguished by chemical structure, have been developed. Four HDACis are FDA approved for the treatment of hematopoietic malignancies: vorinostat, romidepsin, panobinostat, and belinostat. As with MEKi monotherapy, the efficacy of pan-HDACis (vs. selective HDACis) in solid tumors has been lackluster.47
Entinostat, which currently is not approved by the FDA, is a selective benzamide class I HDACi that specifically targets HDAC1 and HDAC3.24 Entinostat simultaneously suppresses protumor regulatory immune responses and stimulates antitumor responses by facilitating antigen presentation and recognition by lymphocytes.48-52 These events fuel an inflammatory TME that inhibits tumor growth. Preclinical data from mouse models of renal, prostate, lung, and colon carcinoma have revealed that entinostat reduces the anti-inflammatory TME and repressed antitumor T-cell activity (mediated by the immunosuppressive functions of myeloid-derived suppressor cells and regulatory T cells) that support tumor growth.49,53 Entinostat also has been found to increase tumor immunogenicity as well as immune recognition and response by upregulating molecules involved in antigen presentation and costimulation.48-52 Preclinical data suggest that entinostat leads to increased gene expression of T-cell costimulatory molecules, chemokines, cytokines, and molecules involved in extravasation, along with elevations in CD8+ T cells and effector memory cells.48,50,51,53,54 Collectively, these events combat tumor growth. Entinostat treatment also enhances antigen presentation via increased major histocompatibility complex class I and II expression.32,50,54 Notably, significant reduction in tumor size with entinostat treatment has been observed only in animal models with an intact immune system, suggesting that the antitumor effect is due to stimulation of an antitumor response by the immune system.48,50
Compared with pan-HDACis, entinostat has a superior safety and efficacy profile because of its selectivity.8 Side effects of entinostat are dose dependent and vary based on administration schedule and patient profile.8 Commonly reported side effects include gastrointestinal upset, anorexia, and hematologic and metabolic abnormalities, and grade 3 and 4 adverse events include hypophosphatemia, anemia, fatigue, neutropenia, and hyponatremia.8,55 However, these events are typically mild and effectively managed with supportive care.8 HDAC activation is implicated in chemotherapy, targeted therapy, and immunotherapy resistance.25 The HDACis show promise in overcoming this resistance. Preclinical data indicate that entinostat pretreatment sensitizes cells to chemotherapy (e.g., decitabine) and kinase inhibitor treatment (e.g., alisertib and axitinib) through an HDAC1-dependent mechanism.56
Combination Therapy: Although in clinical trials HDACi monotherapy demonstrates only modest efficacy for lung cancer, cervical cancer, and melanoma, preclinical data indicate that HDACi administration combined with chemotherapy or immunotherapy enhances the body’s immune response against tumor growth and reduces tumor size.25 Several phase II trials have examined the efficacy of azacitidine, erlotinib, exemestane, or pembrolizumab in combination with entinostat. Limited to no effect was observed with entinostat-erlotinib combination therapy; however, treatment with the entinostat-pembrolizumab combination conferred a modest clinical benefit, with increased sensitivity detected in patients possessing elevated immune markers of antigen presentation.25,55,57 Radiotherapy and ICIs combined with entinostat enhance the antitumor response in lung cancer models by increasing immune-cell infiltration and generating an inflammatory TME.58 Additionally, patients with estrogen receptor–positive breast cancer exhibit increased progression-free survival with entinostat-exemestane combination therapy compared with exemestane alone.59 Other clinical trials examining the efficacy of entinostat combination therapy are currently underway.
Combination Therapy for NSCLC: HDACi combination therapy is gaining traction in the setting of NSCLC.60,61 Namely, cytotoxic and other molecularly targeted therapies in combination with entinostat are emerging as promising treatment regimens.62 Clinical data on trametinib-entinostat combination therapy are limited, but preclinical data are encouraging. A study of NSCLC using a mouse model genetically engineered to harbor the KRAS-LKB1 double mutation found that HDAC3 activity is required for tumor initiation and growth, as evidenced by a marked reduction in tumor growth in mice genetically deficient in HDAC3.63 Compared with vehicle control, neither trametinib nor entinostat alone reduced tumor burden in lung tissue isolated from KRAS-LKB1 double mutant mice; however, trametinib-entinostat combination therapy was found to result in significantly fewer and smaller tumors.63 These findings indicate that entinostat’s tumor-suppressive effect via inhibition of HDAC3 may be especially effective in the presence of MEKi resistance.64
The Oncology Pharmacist’s Role
As medication experts, oncology pharmacists can play a key role in identifying drug-related problems associated with conventional NSCLC treatment options as well as emerging combination therapies for NSCLC, such as trametinib-entinostat, and in educating patients and healthcare providers about these agents’ side-effect profiles, monitoring parameters, and potential drug interactions. The general role of the oncology pharmacist will be discussed here.
Given their expertise regarding cancer medications and associated adverse effects, oncology pharmacists serve as invaluable collaborators on an interdisciplinary healthcare team. Starting with the cancer diagnosis and continuing through the areas of treatment decision making, supportive care, and management of cancer- and treatment-related symptoms, oncology pharmacists’ involvement encompasses the full spectrum of medication-management services.65 Direct patient care is possible when collaborative practice agreements are established, allowing the oncology pharmacist to be fully involved in patient assessment and monitoring as well as treatment selection, administration, and adjustment.
Oncology pharmacists can also take part in developing and implementing medication therapy management programs that improve patients’ quality of life and clinical outcomes. These responsibilities include compiling up-to-date, accurate medication lists; selecting the most suitable therapeutic regimen; monitoring medication effects; identifying potential drug interactions; and monitoring and managing adverse effects. Another major responsibility is to provide patient-directed education and resources to ensure adherence to complex medication regimens in a safe and optimal manner.
Finally, oncology pharmacists play the following indispensable roles: in patient advocacy, by providing optimal and safe care; in the institution or facility, by prioritizing fiscal responsibility; on the healthcare team, by developing educational tools, guidelines, policies, and standards for fellow team members; and in the community, by raising awareness about how individuals can avoid modifiable risk factors for different types of cancer, including NSCLC.
REFERENCES
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72(1):7-33.
2. Tan WW, Huq S. Non-small cell lung cancer (NSCLC). Medscape. https://emedicine.medscape.com/article/279960-overview. Accessed April 16, 2023.
3. Howlader N, Noone AM, Krapcho M, et al, eds. SEER Cancer Statistics Review, 1975-2018. Bethesda, MD: National Cancer Institute. https://seer.cancer.gov/csr/1975_2018/. Accessed April 25, 2023.
4. Ettinger DS, Wood DE, Aisner DL, et al. Non-small cell lung cancer, version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2022;20(5):497-530.
5. Ganti AK, Klein AB, Cotarla I, et al. Update of incidence, prevalence, survival, and initial treatment in patients with non-small cell lung cancer in the US. JAMA Oncol. 2021;7(12):1824-1832.
6. Clark SB, Alsubait S. Non-small cell lung cancer. Treasure Island, FL: StatPearls Publishing; 2023 Jan-.
7. Zhang Y, Meng Q, Sun Q, et al. LKB1 deficiency-induced metabolic reprogramming in tumorigenesis and non-neoplastic diseases. Mol Metab. 2021;44:101131.
8. Connolly RM, Rudek MA, Piekarz R. Entinostat: a promising treatment option for patients with advanced breast cancer. Future Oncol. 2017;13(13):1137-1148.
9. Kirk GD, Merlo C, O’Driscoll P, et al. HIV infection is associated with an increased risk for lung cancer, independent of smoking. Clin Infect Dis. 2007;45(1):103-110.
10. Karampitsakos T, Tzilas V, Tringidou R, et al. Lung cancer in patients with idiopathic pulmonary fibrosis. Pulm Pharmacol Ther. 2017;45:1-10.
11. Tsim S, O’Dowd CA, Milroy R, Davidson S. Staging of non-small cell lung cancer (NSCLC): a review. Respir Med. 2010;104(12):1767-1774.
12. Chevallier M, Borgeaud M, Addeo A, Friedlaender A. Oncogenic driver mutations in non-small cell lung cancer: past, present and future. World J Clin Oncol. 2021;12(4):217-237.
13. Zhao Y, Wang S, Yang Z, et al. Co-occurring potentially actionable oncogenic drivers in non-small cell lung cancer. Front Oncol. 2021;11:665484.
14. Skoulidis F, Heymach JV. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat Rev Cancer. 2019;19(9):495-509.
15. Adderley H, Blackhall FH, Lindsay CR. KRAS-mutant non-small cell lung cancer: converging small molecules and immune checkpoint inhibition. EBioMedicine. 2019;41:711-716.
16. Carretero J, Medina PP, Pio R, et al. Novel and natural knockout lung cancer cell lines for the LKB1/STK11 tumor suppressor gene. Oncogene. 2004;23(22):4037-4040.
17. Shire NJ, Klein AB, Golozar A, et al. STK11 (LKB1) mutations in metastatic NSCLC: prognostic value in the real world. PLoS One. 2020;15(9):e0238358.
18. Ndembe G, Intini I, Perin E, et al. LKB1: can we target an hidden target? Focus on NSCLC. Front Oncol. 2022;12:889826.
19. Stein SC, Woods A, Jones NA, et al. The regulation of AMP-activated protein kinase by phosphorylation. Biochem J. 2000;345 Pt 3(Pt 3):437-443.
20. Vincent EE, Coelho PP, Blagih J, et al. Differential effects of AMPK agonists on cell growth and metabolism. Oncogene. 2015;34(28):3627-3639.
21. Keerthana CK, Rayginia TP, Shifana SC, et al. The role of AMPK in cancer metabolism and its impact on the immunomodulation of the tumor microenvironment. Front Immunol. 2023;14:1114582.
22. Sumbly V, Landry I. Unraveling the role of STK11/LKB1 in non-small cell lung cancer. Cureus. 2022;14(1):e21078.
23. Koivunen JP, Kim J, Lee J, et al. Mutations in the LKB1 tumour suppressor are frequently detected in tumours from Caucasian but not Asian lung cancer patients. Br J Cancer. 2008;99(2):245-252.
24. Shanmugam G, Rakshit S, Sarkar K. HDAC inhibitors: targets for tumor therapy, immune modulation and lung diseases. Transl Oncol. 2022;16:101312.
25. Mamdani H, Jalal SI. Histone deacetylase inhibition in non-small cell lung cancer: hype or hope? Front Cell Dev Biol. 2020;8:582370.
26. Bartling B, Hofmann HS, Boettger T, et al. Comparative application of antibody and gene array for expression profiling in human squamous cell lung carcinoma. Lung Cancer. 2005;49(2):145-154.
27. Hirsch FR, Scagliotti GV, Mulshine JL, et al. Lung cancer: current therapies and new targeted treatments. Lancet. 2017;389(10066):299-311.
28. Yang SR, Schultheis AM, Yu H, et al. Precision medicine in non-small cell lung cancer: current applications and future directions. Semin Cancer Biol. 2022;84:184-198.
29. Vicent S, López-Picazo JM, Toledo G, et al. ERK1/2 is activated in non-small-cell lung cancer and associated with advanced tumours. Br J Cancer. 2004;90(5):1047-1052.
30. Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26(22):3291-3310.
31. Duffy MJ, O’Byrne K. Tissue and blood biomarkers in lung cancer: a review. Adv Clin Chem. 2018;86:1-21.
32. Wang Y, Li N, Jiang W, et al. Mutant LKB1 confers enhanced radiosensitization in combination with trametinib in KRAS-mutant non-small cell lung cancer. Clinical Cancer Res. 2018;24(22):5744-5756.
33. Pawelczyk K, Piotrowska A, Ciesielska U, et al. Role of PD-L1 expression in non-small cell lung cancer and their prognostic significance according to clinicopathological factors and diagnostic markers. Int J Mol Sci. 2019;20(4):824.
34. Stinchcombe TE. Narrative review: blood and tumor biomarker testing in non-small cell lung cancer without an oncogenic driver. Transl Lung Cancer Res. 2023;12(1):158-167.
35. Kim SY, Halmos B. Choosing the best first-line therapy: NSCLC with no actionable oncogenic driver. Lung Cancer Manag. 2020;9(3):LMT36.
36. Kim JH, Kim HS, Kim BJ. Prognostic value of KRAS mutation in advanced non-small-cell lung cancer treated with immune checkpoint inhibitors: a meta-analysis and review. Oncotarget. 2017;8(29):48248-48252.
37. Han J, Liu Y, Yang S, et al. MEK inhibitors for the treatment of non-small cell lung cancer. J Hematol Oncol. 2021;14(1):1.
38. Hainsworth JD, Cebotaru CL, Kanarev V, et al. A phase II, open-label, randomized study to assess the efficacy and safety of AZD6244 (ARRY-142886) versus pemetrexed in patients with non-small cell lung cancer who have failed one or two prior chemotherapeutic regimens. J Thorac Oncol. 2010;5(10):1630-1636.
39. Haura EB, Ricart AD, Larson TG, et al. A phase II study of PD-0325901, an oral MEK inhibitor, in previously treated patients with advanced non-small cell lung cancer. Clin Cancer Res. 2010;16(8):2450-2457.
40. Blumenschein GR Jr, Smit EF, Planchard D, et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC). Ann Oncol. 2015;26(5):894-901.
41. Abdayem P, Planchard D. Ongoing progress in BRAF-mutated non-small cell lung cancer. Clin Adv Hematol Oncol. 2022;20(11):662-672.
42. Tabbò F, Pisano C, Mazieres J, et al. How far we have come targeting BRAF-mutant non-small cell lung cancer (NSCLC). Cancer Treat Rev. 2022;103:102335.
43. Arbour KC, Manchado E, Bott MJ, et al. Phase 1 clinical trial of trametinib and ponatinib in patients with NSCLC harboring KRAS mutations. JTO Clin Res Rep. 2021;3(1):100256.
44. Emery CM, Vijayendran KG, Zipser MC, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A. 2009;106(48):20411-20416.
45. Villanueva J, Infante JR, Krepler C, et al. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep. 2013;4(6):1090-1099.
46. Yu X, Li Y, Jiang G, et al. FGF21 promotes non-small cell lung cancer progression by SIRT1/PI3K/AKT signaling. Life Sci. 2021;269:118875.
47. Ceccacci E, Minucci S. Inhibition of histone deacetylases in cancer therapy: lessons from leukaemia. Br J Cancer. 2016;114(6):605-611.
48. Truong AS, Zhou M, Krishnan B, et al. Entinostat induces antitumor immune responses through immune editing of tumor neoantigens. J Clin Invest. 2021;131(16):e138560.
49. Shen L, Pili R. Class I histone deacetylase inhibition is a novel mechanism to target regulatory T cells in immunotherapy. Oncoimmunology. 2012;1(6):948-950.
50. Smith HJ, McCaw TR, Londono AI, et al. The antitumor effects of entinostat in ovarian cancer require adaptive immunity. Cancer. 2018;124(24):4657-4666.
51. Sidiropoulos DN, Rafie CI, Jang JK, et al. Entinostat decreases immune suppression to promote antitumor responses in a HER2+ breast tumor microenvironment. Cancer Immunol Res. 2022;10(5):656-669.
52. Wang X, Waschke BC, Woolaver RA, et al. HDAC inhibitors overcome immunotherapy resistance in B-cell lymphoma. Protein Cell. 2020;11(7):472-482.
53. Orillion A, Hashimoto A, Damayanti N, et al. Entinostat neutralizes myeloid-derived suppressor cells and enhances the antitumor effect of PD-1 inhibition in murine models of lung and renal cell carcinoma. Clin Cancer Res. 2017;23(17):5187-5201.
54. Hicks KC, Knudson KM, Lee KL, et al. Cooperative immune-mediated mechanisms of the HDAC inhibitor entinostat, an IL15 superagonist, and a cancer vaccine effectively synergize as a novel cancer therapy. Clin Cancer Res. 2020;26(3):704-716.
55. Hellmann MD, Jänne PA, Opyrchal M, et al. Entinostat plus pembrolizumab in patients with metastatic NSCLC previously treated with anti-PD-(L)1 therapy. Clin Cancer Res. 2021;27(4):1019-1028.
56. Hess L, Moos V, Lauber AA, et al. A toolbox for class I HDACs reveals isoform specific roles in gene regulation and protein acetylation. PLoS Genet. 2022;18(8):e1010376.
57. Witta SE, Jotte RM, Konduri K, et al. Randomized phase II trial of erlotinib with and without entinostat in patients with advanced non-small-cell lung cancer who progressed on prior chemotherapy. J Clin Oncol. 2012;30(18):2248-2255.
58. Kim Y, Park K, Kim YJ, et al. Immunomodulation of HDAC inhibitor entinostat potentiates the anticancer effects of radiation and PD-1 blockade in the murine Lewis lung carcinoma model. Int J Mol Sci. 2022;23(24):15539.
59. Yardley DA, Ismail-Khan RR, Melichar B, et al. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J Clin Oncol. 2013;31(17):2128-2135.
60. Topper MJ, Vaz M, Chiappinelli KB, et al. Epigenetic therapy ties MYC depletion to reversing immune evasion and treating lung cancer. Cell. 2017;171(6):1284-1300.e21.
61. Adeegbe DO, Liu Y, Lizotte PH, et al. Synergistic immunostimulatory effects and therapeutic benefit of combined histone deacetylase and bromodomain inhibition in non-small cell lung cancer. Cancer Discov. 2017;7(8):852-867.
62. Ruiz R, Raez LE, Rolfo C. Entinostat (SNDX-275) for the treatment of non-small cell lung cancer. Expert Opin Investig Drugs. 2015;24(8):1101-1109.
63. Eichner LJ, Curtis SD, Brun SN, et al. HDAC3 is critical in tumor development and therapeutic resistance in Kras-mutant non-small cell lung cancer. Sci Adv. 2023;9(11):eadd3243.
64. Kun E, Tsang YTM, Ng CW, et al. MEK inhibitor resistance mechanisms and recent developments in combination trials. Cancer Treat Rev. 2021;92:102137.
65. Holle LM, Boehnke Michaud L. Oncology pharmacists in health care delivery: vital members of the cancer care team. J Oncol Pract. 2014;10(3):e142-e145.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
To comment on this article, contact rdavidson@uspharmacist.com.



