US Pharm. 2007;32(3):HS-35-HS-48.
Thromboembolism is one of the leading causes
of morbidity and mortality in the United States. Venous thromboembolism (VTE)
most frequently presents as either deep venous thrombosis (DVT) or pulmonary
embolism, and these disorders often occur in patients who experience trauma,
undergo major surgery, are immobilized for an extended period of time, or have
a hypercoagulable disorder.1-5 Arterial thrombosis is less common
and may lead to unstable angina, myocardial infarction, stroke, and peripheral
arterial disease.3
Thrombosis, or blood clotting, is the
terminal product of a complex cascade of spontaneous enzymatic reactions
involving platelet aggregation and the body's coagulation system. A thrombus
can occur when the extrinsic portion of the body's clotting cascade is
activated by endothelial damage, which leads to the release of large amounts
of tissue factor. This creates a complex that activates factor VII. The tissue
factor-VIIa activates factor X. Factor Xa binds with factor Va on the
phospholipid surface of platelets and forms the prothrombinase complex. In the
presence of calcium, the prothrombinase complex converts factor II
(prothrombin) to factor IIa (thrombin).6-10
Through positive feedback mechanisms,
thrombin amplifies its own production and activates factors V, VIII, and XI.
6,9-10 Activation of factor XI modulates production of more thrombin
through the intrinsic clotting cascade. Factor XIa and the tissue factor VIIa
complex activate factor IX. Factor IXa and the thrombin-activated factor VII
form the tenase complex on the phospholipid surface of platelets. In
combination with calcium, the tenase complex increases the formation of factor
Xa. With the increased production of factors Xa and Va by thrombin, thrombin
production is accelerated.6,8-10 Thrombin also interacts with
thrombomodulin to produce activated protein C. In an attempt to reduce or
control thrombin production, activated protein C, with its cofactor protein S,
deactivates factors Va and VIIIa. In addition, thrombin is responsible for the
cleavage of fibrinogen in insoluble fibrin and activates factor XIII to
stabilize the fibrin meshwork in the thrombus. Two central components of this
complex clotting cascade are factors Xa and IIa. Blockade of factor X, in
midstream of the clotting cascade, blocks the progression of the cascade to
form thrombin. Blockade of factor II, one of the terminal reactions in the
formation of a thrombus, prevents the formation of fibrin.8
Drugs whose mechanisms of action block the
above coagulation pathways have the potential to effectively prevent and/or
treat thromboembolic disease. Thromboembolic disease can be a debilitating and
potentially fatal disorder, and it is very important to treat it quickly and
aggressively.11-15 The purpose of this article is to provide the
pharmacist with an overview of factor Xa inhibitors and direct thrombin
inhibitors (DTIs) utilized in the U.S.
DIRECT FACTOR Xa INHIBITORS
Direct factor Xa inhibitors bind
to and inhibit factor Xa without a requirement for antithrombin III (AT III).
16 Their activity is specific for factor Xa, with no effect on other
components of the coagulant cascade. The major advantage of these agents lies
in their small size and their ability to inactivate circulating, as well as
bound, forms of factor Xa. This inhibition occurs in a stoichiometric
manner--one molecule of direct factor Xa inhibitor inactivates one molecule of
factor Xa. Some direct factor Xa inhibitors are currently under development in
preclinical stages, but none are currently approved for use in the U.S.
17,18
INDIRECT FACTOR XaINHIBITORS
Introduced to the market in
December 2001, fondaparinux is the only drug available in a new class of drugs
known as pentasaccharides.19 The development of this small,
completely synthetic molecule was based on the native pentasaccharide sequence
in unfractionated heparin (UFH).18 Because fondaparinux is produced
by chemical synthesis, it has batch-to-batch consistency, which eliminates the
risk of pathogen contamination associated with animal-sourced agents such as
heparins. Fondaparinux specifically targets factor Xa and exerts its
antithrombotic effect by inhibiting thrombin generation and, therefore, fibrin
formation. Unlike heparins, which contain antifactor IIa activity in varying
degrees, fondaparinux has no effect on circulating thrombin's regulatory
function in the control of hemostasis.20-22
Fondaparinux
Pharmacotherapeutics:
Fondaparinux is closely related in chemical structure and function to heparin
and low-molecular-weight heparins (LMWHs). The molecule is a copy of the AT
III binding area of the heparin compound. Fondaparinux is approved only for
the prevention of VTE following total hip fracture surgery, hip replacement
surgery, or knee replacement surgery. It is currently under evaluation for the
treatment of unstable angina and acute coronary syndromes. Fondaparinux is
available in a strength of 2.5 mg/0.5 mL in single-dose, prefilled syringes.
It is administered by subcutaneous (SC) injection into fatty tissue at
alternating injection sites (e.g., between the left and right anterolateral or
posterolateral abdominal wall).
Pharmacokinetics: Fondaparinux
is a fixed-dose antithrombotic agent that is administered by SC injection once
daily, starting six to eight hours after surgery. It does not require routine
anticoagulation monitoring for therapeutic effect.19,23,24 It has a
predictable pharmacokinetic profile and a dose-response relationship.
Following SC injection, it is rapidly and completely (100%) absorbed into
plasma, with a peak concentration occurring in approximately two to three
hours. Fondaparinux has an elimination half-life of 17 to 21 hours, with an
average half-life of 13 hours in patients with a creatinine clearance (CrCl)
of 90 to 140 mL/min, 29 hours in those with a CrCl of 31 to 60 mL/min, and 72
hours in those with a CrCl of 10 to 30 mL/min.13,25,26 It is
primarily eliminated unchanged in the urine in patients with normal renal
function. In healthy patients as old as 75 years, up to 77% of the
administered dose is eliminated unchanged in the urine in 72 hours. In
patients with renal insufficiency, the half-life is prolonged.27
Pharmacodynamics: Fondaparinux
binds to AT III and potentiates its antifactor Xa activity (i.e., acts as an
AT III catalyst). It reversibly binds to antithrombin, causing a
conformational change that potentiates its antifactor Xa activity by a factor
of 300.19,22-25,28,29 These agents may provide a more efficient
mechanism for the control of fibrin formation, since inactivating one molecule
of factor Xa by AT III inhibits the generation of 50 thrombin molecules.30
If low levels of antithrombin exist, the
activity of fondaparinux is reduced. This agent selectively inhibits thrombin
production indirectly (by inactivating factor Xa). Fondaparinux's lack of
activity on circulating thrombin levels is due to its small molecular size,
which allows it to bind to factor Xa but not to the thrombin molecule. Unlike
heparins, fondaparinux does not affect platelet function, nor does it inhibit
platelet aggregation stimulated by various agonists.26,31,32
Fondaparinux does not react with heparin platelet factor 4 antibodies, thus
potentially eliminating the risk of heparin-induced thrombocytopenia (HIT).
Fondaparinux inactivates circulating, as well as bound, forms of factor Xa.
Interestingly, the fondaparinux molecule is not consumed in the reaction with
factor Xa. Once AT III binds to factor Xa, fondaparinux is released to
interact with other AT III molecules.
Fondaparinux has no effect on prothrombin
time (PT), activated partial thromboplastin time (aPTT), or bleeding-time lab
test results. Patients should be monitored periodically with routine complete
blood counts (CBCs) with platelets, serum creatinine level, and stool occult
blood tests. If platelet counts fall below 100,000 mm3,
fondaparinux should be discontinued.
Overdoses of fondaparinux cannot be treated
with protamine sulfate, which can be used for overdoses of UFH or LMWH. There
is no known antidote for overdoses of fondaparinux.
Adverse Effects:19,23,24
The most common adverse effects are bleeding (major 2.7%, minor 3%) and
secondary local bruising. In clinical trials, the incidence of bleeding
reportedly is doubled in patients weighing less than 50 kg compared with
patients weighing 50 kg or more (5.4% vs 2.1%).12,19,31
Fondaparinux does not promote immune-mediated HIT, although it can cause
reversible low platelet counts.
Contraindications and Precautions:
19,23,24 Fondaparinux is contraindicated in patients who weigh less than
50 kg (clearance is reduced by 30%) or have severe renal impairment (i.e.,
CrCl <30 mL/min), active major bleeding, thrombocytopenia associated with a
positive in vitro test for antiplatelet antibodies in the presence of
fondaparinux, or uncontrolled hypertension.
In patients undergoing anesthesia using an
epidural or spinal catheter, fondaparinux can cause spinal or epidural
hematoma, which can result in permanent paralysis. Fondaparinux is not
interchangeable with heparin or heparinoids on a unit-for-unit or mg-to-mg
basis.1
Fondaparinux is not recommended for use in
pediatric patients. It is a pregnancy category B drug.
Drug Interactions:19,23,24
Platelet aggregation inhibitors, anticoagulants, thrombolytics, nonsteroidal
anti-inflammatory drugs, and other drugs and herbal products that interfere
with normal hemostasis should be avoided. Fondaparinux produces less than 30%
inhibition of CYP2A6-mediated warfarin metabolism and minimal effects on
CYP1A2, CYP2C19, CYP2D6, CYP3A4, and CYP3E. Due to the lack of hepatic
metabolism, fondaparinux is unlikely to cause major drug interactions mediated
by these CYP450 enzymes.12
DIRECT THROMBIN INHIBITORS
In 1884, John Haycraft identified
the anticoagulant in the saliva of the European medicinal leech, which he
named hirudin. Until the discovery of heparin in 1916, hirudin was the
only means of preventing blood clots. In the late 1950s, the active ingredient
in hirudin was identified as a peptide structure containing 65 amino acids,
with a calculated molecular weight of about 7,000 Da. In 1976, the primary
chemical structure of hirudin was determined; this structure is the prototype
compound for DTIs.12,33
Although native hirudin is not commercially
available in the U.S., due to the large amount of leeches needed to obtain
hirudin, derivatives have been produced by recombinant DNA technologies,
including point mutations and N-terminal modifications. Clotting assay
studies have shown that the anticoagulant activity of synthetic hirudin is as
high as that of native hirudin.
DTIs are the newest group of potent
anticoagulants. The first commercially available DTI product in the U.S.,
lepirudin, was introduced in March 1998. In addition to lepirudin, three other
parenterally administered DTI products are currently available: argatroban,
introduced in June 2000; bivalirudin, introduced in December 2000; and
desirudin, introduced in April 2003.
Because DTIs do not bind to the
fibrin-binding site, they can bind both unbound and fibrin-bound thrombin,
preventing the dual processes of thrombus initiation and propagation. In
contrast, heparin-activated antithrombin binds to the active site of thrombin
but also blocks the fibrin-binding site. Thus, when thrombin and fibrin are
already bound, which occurs within a fibrin clot, heparin is unable to
inactivate thrombin.34 Based on these pharmacologic properties,
DTIs appear to be more potent than traditional anticoagulants. In addition,
they are not inhibited by platelet factor 4 or associated with the development
of HIT. Although there are a number of zymogens in the coagulation cascade,
thrombin is central because it is the terminal precursor to fibrin formation.
In addition, thrombin is able to amplify its own production; thus, it is a
natural target for pharmacologic intervention.
Bivalirudin
Pharmacotherapeutics:
Bivalirudin, formerly known as hirulog, is a semisynthetic 20-amino acid
polypeptide (2,178 Da) analog of recombinant hirudin. Bivalirudin is indicated
for use in conjunction with 325 mg of aspirin for patients with unstable
angina who are undergoing percutaneous transluminal coronary angioplasty. It
may also be used off label in patients with HIT. It is administered by
intravenous (IV) injection and IV infusion. For patients with a CrCl of
greater than 60 mL/min, no reduction in dosage is needed. However, for
patients with a CrCl of 30 to 50 mL/min or 10 to 29 mL/min, dosage should be
reduced by 20% and 60%, respectively. For dialysis-dependent patients (off
dialysis), dosage should be reduced by 90%.19,23
Pharmacokinetics: When
bivalirudin is administered intravenously, bivalirudin exhibits linear
pharmacokinetics and pharmacologic effects. It does not bind to red blood
cells or plasma proteins other than thrombin. It is cleared from plasma by a
combination of renal and proteolytic cleavage. The half-life of bivalirudin in
patients with normal renal function is 25 minutes; and in patients with a CrCl
of 10 to 29 mL/min or in those on hemodialysis, the half-life is 57 minutes
and 3.5 hours, respectively. Coagulation times return to baseline values in
approximately one hour following discontinuation of IV infusion.19,23
Pharmacodynamics:19,23
Bivalirudin acts immediately as a specific and reversible DTI, binding to
circulating and clot-bound thrombin. It is not dependent upon AT III as a
cofactor and has no platelet aggregation response. DTIs target sites on the
thrombin molecule responsible for substrate recognition and/or cleavage.35
The substrate recognition site (exosite 1) acts as a docking station, binding
thrombin to fibrinogen prior to its enzymatic actions. The catalytic site
(active site) is responsible for the enzymatic actions of thrombin, including
activation of platelets and cleavage of fibrinogen for thrombus formation.
36 By blocking either the active site alone or both the active site and
the exosite 1, DTIs specifically inhibit thrombin activity.
For patients with renal impairment,
activated clotting time and CBC should be monitored. In addition, bivalirudin
prolongs aPTT, PT, and thrombin time (TT) lab test values.
It is important to note that discontinuation of
bivalirudin leads to a reduction in anticoagulant effects. Its short half-life
allows for watchful waiting. Patients should be monitored carefully for signs
of bleeding. There is no known antidote for bivalirudin overdose, but
hemodialysis will help remove the drug.
Contraindications and Precautions:
19,23 In general, concurrent use of other anticoagulants and platelet
aggregation inhibitors, except aspirin, should be avoided. No harm to the
fetus has been demonstrated in animal studies, but safety and efficacy for use
in pregnant women have not been established. Since concurrent use of
bivalirudin and aspirin may lead to maternal or fetal adverse effects,
especially during the third trimester, aspirin should be used only if its
benefits outweigh the risks. Bivalirudine is a pregnancy category B drug.
Nausea may increase with concurrent use of
selective serotonin reuptake inhibitors, lithium, or valproate. Bivalirudin is
not intended for intramuscular (IM) administration, and all other IM
injections should be avoided.
Adverse Effects:19,23
The most serious adverse effects of bivalirudin are minor and major bleeding
(~4%). Hemorrhage may occur at virtually any site. Other adverse effects
include hypotension (12%), headache (12%), nausea (15%), back pain (42%),
hypertension (6%), insomnia (7%), and anxiety (6%). Adverse effects with an
incidence rate of approximately 5% include fever, nervousness, vomiting,
dyspepsia, abdominal pain, pelvic pain, and bradycardia.
Drug Interactions:19,23,37
Coadministration of bivalirudin with ticlopidine, abciximab, heparin, and
LMWH will likely increase the risk of major bleeding. The safety and efficacy
of bivalirudin have not been established when used in conjunction with
platelet aggregation inhibitors other than aspirin (i.e., glycoprotein
IIb/IIIa inhibitors).
Lepirudin
Pharmacotherapeutics:
19,23 Lepirudin (6,980 Da) is a recombinant analog of hirudin, a
65-amino acid polypeptide. It is indicated for the treatment of
heparin-associated thrombocytopenia and associated thromboembolic disease.
Lepirudin (rDNA) is a recombinant hirudin derived from yeast cells.
Unlabeled uses include adjunct therapy for
treatment of unstable angina and acute myocardial infarction without ST
elevation, for prevention of DVT, and in patients undergoing percutaneous
coronary interventions.
Lepirudin is administered as a slow IV bolus
(0.4 mg/kg [up to 110 kg] over 15 to 20 seconds), followed by continuous IV
infusion (0.15 mg/kg [up to 110 kg/hour]). The maximum initial bolus is 44 mg,
and the maximum initial infusion dose is 16.5 mg/hour.
When dosing with a thrombolytic (e.g.,
alteplase, urokinase, or streptokinase) an initial IV bolus of 0.2 mg/kg
should be administered, followed by continuous IV infusion (0.1 mg/kg).
Pharmacokinetics:19,23
Following IV administration, distribution of lepirudin is essentially
confined to the body's extracellular fluids. It is thought to be metabolized
by release of amino acids via catabolic hydrolysis of the parent drug. It has
a half-life of approximately 10 minutes. Approximately 48% of the administered
dose is excreted in the urine, and about 35% is unchanged along with other
fragments of the parent drug.
Pharmacodynamics:19,23
Lepirudin irreversibly binds specifically to circulating and clot-bound
thrombin. Its inhibition of thrombin occurs independently of AT III and
heparin cofactor III and has no direct effect on platelet function, except
that it inhibits thrombin-induced platelet activation. Lepirudin is a highly
specific DTI. The mechanism of action of lepirudin is independent of
antithrombin, and it is not inhibited by platelet factor 4. One molecule of
lepirudin binds to one molecule of thrombin to block its thrombogenic
activity. One antithrombin unit (ATU) is the amount of lepirudin that
neutralizes one unit of World Health Organization preparation 89/588 of
thrombin. The activity of lepirudin is approximately 16,000 ATU/mg.
Lepirudin therapy should be monitored daily
using the aPTT ratio (i.e., patient's aPTT at a given time over an aPTT
reference value, usually mean of the normal laboratory range for aPTT) in
patients with an increased risk of bleeding and/or renal impairment. The
target aPTT ratio or therapeutic window during treatment should be 1.5 to 2.5.
The aPTT test value increases in a dose-dependent fashion. The patient's
baseline aPTT should be determined prior to initiating lepirudin therapy. If
the baseline aPTT ratio is 2.5 or greater, lepirudin therapy should not be
initiated. If the aPTT ratio is greater than 2.5, stop IV infusion for two
hours. If therapy is restarted, decrease the infusion by 50% (no additional
bolus should be administered) and determine the aPTT ratio again four hours
later. If the aPTT ratio is less than the target range, increase the infusion
rate in increments of 20% and determine the aPTT ratio again in four hours.
The TT is not a suitable test for routine monitoring of lepirudin therapy.
Discontinuation of lepirudin leads to a
reduction in anticoagulant effects. Its short half-life allows for watchful
waiting. Patients should be monitored closely for signs of bleeding. There is
no known antidote for lepirudin, but hemodialysis will help remove the drug.
Contraindications and Precautions:
19,23 Active major bleeding; known allergic or hypersensitivity
reactions to lepirudin, including anaphylactic reactions.
Adverse Effects:19,23
The most common hemorrhagic adverse effects include hemorrhage from puncture
sites and wounds (10.6%), anemia (12.4%), and other hematomas and unclassified
bleeding (10.6%). Nonhemorrhagic adverse effects include abnormal liver
function (5.3%), fever (4.4%), and pneumonia (4.4%).
Drug Interactions:19,23,37
Concomitant treatment with thrombolytics and/or warfarin may increase the
risk of bleeding.
Argatroban
Pharmacotherapeutics:19,23
Argatroban (527 Da) is a synthetic N2-substituted arginine derivative from l
-arginine. It is indicated for prophylaxis or treatment of thrombosis in
patients with HIT or HIT with thrombosis.
Argatroban is administered by continuous IV
infusion. In patients with normal hepatic function, the recommended initial IV
infusion rate is 2 mcg/kg/min. In patients with liver dysfunction, the initial
infusion rate is 0.5 mcg/kg/min. The dosage is adjusted to attain a
steady-state aPTT of 1.5 to three times the mean normal value or baseline
value.19,23,37,38
Pharmacokinetics:19,23,39
Argatroban is rapidly metabolized in the liver via hydroxylation and
aromatization.38 The metabolized products are removed via biliary
excretion, so dosing reductions and careful monitoring are recommended in
patients with hepatic dysfunction.40 The drug has an elimination
half-life of about 50 minutes, and renal impairment has no influence on the
elimination half-life. When infusion of argatroban is discontinued, the aPTT
value returns to baseline within two to four hours.
Pharmacodynamics:19,23,39
Argatroban is a selective thrombin inhibitor that binds reversibly to the
active site of the thrombin molecule.41 It does not require the
cofactor antithrombin. It exerts its anticoagulant effects by inhibiting
thrombin-catalyzed or -induced reactions, including fibrin formation;
activation of coagulation factors V, VIII, and XIII; protein C formation; and
platelet aggregation. It does not interact with heparin-induced antibodies.
The aPTT lab test is used to monitor argatroban therapy and its anticoagulant
activity.42
Discontinuation of argatroban leads to a
reduction in anticoagulant effects. Its short half-life allows for watchful
waiting. Closely monitor the patient for signs of bleeding. There is no known
antidote for argatroban.
Contraindications and Precautions:
19,23,39 Overt major bleeding; known allergy or hypersensitivity
reactions to argatroban.
Adverse Effects:19,23,39
The most common adverse effect of argatroban is hemorrhage. About 12% of
patients experience hematuria. Allergic reactions (i.e., dyspnea, cough, rash)
occur in 10% of patients, particularly in those receiving either thrombolytic
drugs (i.e., strepto kinase) or contrast media for coronary angioplasty.
Drug Interactions:19,37,39
Coadministration of argat roban with antiplatelet agents,
thrombolytics, and other anticoagulants will likely increase the risk of
bleeding.
Desirudin
Pharmacotherapeutics:19,23,39
Desirudin is a recombinant hirudin that is being investigated for the
prophylaxis of DVT, which may lead to pulmonary embolism, in patients
undergoing elective hip replacement surgery.
The suggested initial dosage is 15 mg every
12 hours administered by deep SC injection, with the initial dose given up to
five to 15 minutes before surgery but after induction of regional block
anesthesia (if used). Average duration of desirudin treatment is nine to 12
days. Initiate dosage in patients with moderate renal insufficiency (i.e.,
CrCl = 31 to 60 mL/min) is 5 mg every 12 hours. In patients with severe renal
insufficiency (i.e., CrCl <31 mL/min), the initial dosage is 1.7 mg every 12
hours. Desirudin is available as 15.75-mg vials and must be reconstituted with
0.5 mL of manitol ([3%] in water for injection) prior to administration.
Desirudin cannot be used interchangeably with other hirudin products.
Pharmacokinetics:19,23,39
Following SC administration, desirudin is completely absorbed. Plasma
concentrations are dose proportional, with the maximum level and area under
the curve occurring between one and three hours. Desirudin binds specifically
and directly to thrombin, forming an extremely tight, noncovalent complex.
Desirudin is metabolized by the liver and excreted by the kidney. Total
urinary excretion of unchanged desirudin amounts to 40% to 50% of the
administered dose. The mean terminal elimination half-life is approximately
two to three hours.
Pharmacodynamics:19,23,39
Desirudin is a highly specific DTI. It selectively inhibits free circulating
and clot-bound thrombin. One molecule of desirudin binds to one molecule of
thrombin. The pharmacodynamic effect of desirudin is measured by an aPTT lab
test. Patients with increased risk of bleeding and/or renal impairment should
be monitored daily using this test.
No specific antidote for desirudin is
available. Its short half-life allows for watchful waiting. The anticoagulant
effect is partially reversed using thrombin-rich plasma concentrates. The aPTT
level can be reduced by IV administration of desmopressin.
Contraindications and Precautions:
19,23,39 Desirudin is contraindicated in patients with known
hypersensitivity to natural or recombinant hirudin and in patients with active
bleeding and/or irreversible coagulation disorders.
This agent should be used with caution in
patients with a high risk of hemorrhage and in those with spinal/epidural
anesthesia, moderate-to-severe renal function impairment (i.e., CrCl <60
mL/min), and hepatic function impairment. Caution should be taken when
treating elderly patients, pregnant (pregnancy category C) or lactating
females, and children. Desirudin should also be used with caution in patients
switching from oral anticoagulants to desirudin or from desirudin to oral
anticoagulants and in patients with antibodies against or reexposure to
hirudin.
Adverse Effects: The most
common adverse effect is hemorrhage.
Drug Interactions:19,23,39
Certain agents (i.e., dextran 40, systemic glucocorticoids, thrombolytics,
and anticoagulants) may enhance the risk of hemorrhage.
Role of the Pharmacist
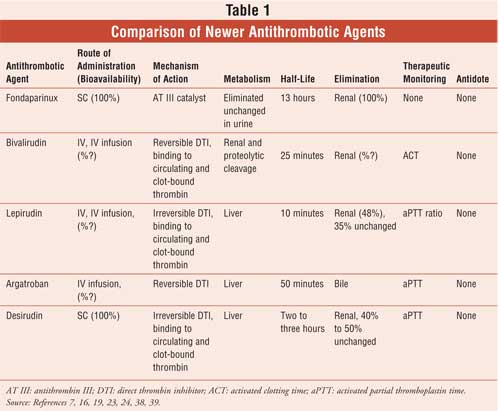
As the most accessible health care professional, the pharmacist can have a significant role in assisting outpatients who are concerned about avoiding adverse effects when taking newer anticoagulant agents. In a collaborative practice environment (i.e., hospitals or clinic settings), the pharmacist can work closely with both patients and physicians to monitor the antithrombotic therapy of inpatients or outpatients. To accomplish this, pharmacists should be familiar with newer anticoagulant drugs (see Table 1) and common adverse effects associated with the use of these agents. The pharmacist should also be able to appropriately educate patients and/or their family members on the goals of therapy, living with a thromboembolic disease, and precautions to follow while taking an antithrombotic drug. In some instances, avoiding some very troublesome drugs can help prevent clinically significant drug interactions and avoid emergency room visits.
Conclusions
Much of the published clinical data indicate that
newer anticoagulants are at least as effective as traditional anticoagulants,
and some are more effective (i.e., fondaparinux in orthopedic surgery, hirudin
in acute coronary syndromes).43 DTIs have the potential to decrease
bleeding risk, compared with traditional anticoagulants in some circumstances
(e.g., bivalirudin in patients given percutaneous intervention).
Oral formulations of DTIs are not currently
available but are in developmental phases. Oral agents showing promise have
the potential to revolutionize antithrombotic therapy, allowing for safer and
more convenient administration, as well as equal or superior therapeutic
effectiveness. However, to date, warfarin is the only oral agent available in
the U.S. for chronic antithrombotic therapy.
References
1. 2002 heart and stroke
statistical update. Dallas, Tex: American Heart Association; 2003.
2. Anderson FA Jr, Wheeler HB,
Goldberg RJ, et al. A population-based perspective of the hospital incidence
and case-fatality rates of deep vein thrombosis and pulmonary embolism. The
Worcester DVT Study. Arch Intern Med. 1991;151:933-938.
3. Tavazzi L. Clinical epidemiology
of acute myocardial infarction. Am Heart J. 1999;138:S48-S54.
4. Virchow RLK. Thrombosis and
Emboli (1846-1856). Science History Publications. Canton, Mass: Watson
Publishing International; 1998.
5. Federman DG, Kirsner RS. An update
on hypercoagulable disorders. Arch Intern Med. 2001;161:1051-1056.
6. Colman RW, Clowes AW, George JH,
et al. Overview of Homeostasis and Thrombosis: Basic Principles and
Clinical Practice. 4th ed. Philadelphia, Pa: Lippincott Williams &
Wilkins; 2001:3-16.
7. Haines ST, Bussey HI. Thrombosis
and the pharmacology of antithrombotic agents. Ann Pharmacother.
1995;29:892-905.
8. Dahlback B. Blood coagulation.
Lancet. 2000;355:1627-1632.
9. Ginsberg MH, Du X, O'Toole TE,
Loftus JC. Platelet integrins. Thromb Haemost. 1995;74:352-359.
10. Harker LA, Mann KG. Thrombosis
and fibrinolysis. In: Verasteraete M, Fuster V, Topol EJ, eds.
Cardiovascular Thrombosis. 2nd ed. Philadelphia, Pa: Lippincott-Raven;
1998:3-22.
11. Hirsh J, Hoak J. Management of
deep vein thrombosis and pulmonary embolism. A statement for health care
professionals. Council on Thrombosis (in consultation with the Council on
Cardiovascular Radiology), American Heart Association. Circulation.
1996;93:2212-2245.
12. Lensing AW, Prandoni P, Prins MH,
Buller HR. Deep-vein thrombosis. Lancet. 1999;353:479-485.
13. Goldhaber SZ. Pulmonary embolism.
N Engl J Med. 1998;339:93-104.
14. Haines ST, Bussey HI. Diagnosis
of deep vein thrombosis. Am J Health Syst Pharm. 1997;54:66-74.
15. Prandoni P, Lensing AW, Prins MR.
The natural history of deep-vein thrombosis. Semin Thromb Hemost.
2000;23:185-188.
16. Turpie AG, Gallus AS, Hoek JA. A
synthetic pentasaccharide for the prevention of deep-vein thrombosis after
total hip replacement. N Engl J Med. 2001;344:619-625.
17. Herbert JM, Bernat A, Dol F, et
al. DX 9065A, a novel, synthetic, selective and orally active inhibitor of
factor Xa: in vitro and in vivo studies. J Pharmacol Exp Ther.
1996;276:1030-1038.
18. Herbert JM, Petitou M, Lormeau
JC. SR90107A/Org 31540, a novel anti-factor Xa antithrombotic agent.
Cardiovasc Drug Rev. 1997;15:1-26.
19. Kastrup EK. Drug Facts and
Comparisons. St. Louis, Mo: Facts and Comparisons (Firm); 2005.
20. Choay J, Petitou M, Lormeau JC,
et al. Structure-activity relationship in heparin: a synthetic pentasaccharide
with high affinity for antithrombin III and eliciting high anti-factor Xa
activity. Biochem Biophys Res Commun. 1983;116:492-499.
21. van Boeckel CA, Beetz T, Vos JN,
et al. Synthesis of a pentasaccharide corresponding to the antithrombin III
binding fragment of heparin. J Carbohydr Chem. 1985;4:293–321.
22. Petitou M, Duchaussoy P, Lederman
I, et al. Synthesis of heparin fragments: a methyl alpha-pentaoside with high
affinity for antithrombin III. Carbohydr Res. 1987;167:67-75.
23. Anderson PO, Knoben JE, Troutman
WG. Handbook of Clinical Drug Data. 10th ed. New York, NY: McGraw-Hill;
2002.
24. Arixtra package
literature. Organon Sanofi-Synethlabo LLC; December 2001.
25. Toglia MR, Nolan TE. Venous
thromboembolism during pregnancy: a current review of diagnosis and
management. Obstet Gynecol Surv. 1997;52:60-72.
26. McFarlane RG. An enzyme cascade
in the blood clotting mechanism, and its function as a biochemical amplifier.
Nature. 1964;202:498-499.
27. Faaij RA, et al. The influence of
renal function on the pharmacokinetics and pharmacodynamics of
ORG31540/SR90107A. Thromb Haemost. 1997;76(suppl):PS379.
28. Choay J, Petitou M, Lormeau JC,
et al. Structure-activity relationship in heparin: a synthetic pentasaccharide
with high affinity for antithrombin III and eliciting high anti-factor Xa
activity. Biochem Biophys Res Commun. 1983;116:492-499.
29. Van Boeckel CA, Beetz T, Vos JN,
et al. Synthesis of a pentasaccharidce corresponding to the antithrombin III
binding fragment of heparin. J Carbohydr Chem. 1985;4:293-321.
30. Wessler S, Yin ET. On the
antithrombotic action of heparin. Thromb Diath Haemorrh. 1974;32:71-78.
31. Nemerson Y. Tissue factor and
hemostasis. Blood. 1988;71:1-8.
32. Davie EW, Fujikawa K, Kisiel W.
The coagulation cascade initiation, maintenance, and regulation.
Biochemistry. 1991;30:10363-10370.
33. Agnelli G, Sonagila F. Clinical
status of direct thrombin inhibitors. Crit Rev Oncol Hematol.
1993;31:91-117.
34. Link KP. The discovery of
dicumarol and its sequels. Circulation. 1959;19:97-107.
35. Lefkovits J, Topol EJ. Direct
thrombin inhibitors in cardiovascular medicine. Circulation.
1994;90:1522-1536.
36. Samama MM, Kher A.
Anticoagulation: the old and the new. Hamostaseologie. 1998;18(suppl
A):S27-S32.
37. Kastrup EK. Drug Interactions
Facts. St. Louis, Mo: Facts and Comparisons (Firm); 2004.
38. Swan SK, Hursting MJ. The
pharmacokinetics and pharmacodynamics of argat roban: effects of age,
gender, and hepatic or renal dysfunction. Pharmacotherapy.
2000;20:318-329.
39. Haines ST, Bussy SI. Thrombosis
and the pharmacology of antithrombotic agents. Ann Pharmacother.
1995;29:892-905.
40. GlaxoSmithKline and Texas
Biotechnology receive FDA approval for new indication for argatroban.
Available at: www.argatroban.com. Accessed January 8, 2003.
41. Hursting MJ, Alford KL, Becker
JC, et al. Novastan (brand of argatroban): a small-molecule, direct thrombin
inhibitor. Semin Thromb Hemost. 1997;23:503-516.
42. Kondo LM, Wittkowsky AK, Wiggins
BS. Argatroban for prevention and treatment of thromboembolism in
heparin-induced thrombocytopenia. Ann Pharmacother. 2001;35:440-451.
43. Brown CH. An overview of
traditional anticoagulants. USPharm. 2006;2:HS6-HS24.
To comment on this article, contact
editor@uspharmacist.com.


