US Pharm. 2019;44(5)(Specialty&Oncology suppl):4-8.
ABSTRACT: Acute myeloid leukemia (AML) represents a heterogeneous group of acute hematologic malignancies produced by the myeloid lineage within the hematopoietic system. Recent advances in molecular genetics have led to the discovery of new oncogenic signaling pathways and molecular or genetic aberrations involved in the malignant transformation of myeloid precursors to leukemic blasts, which in turn has led to improved diagnostic and therapeutic strategies for patients with AML. The frequent occurrence of mutations and gene alterations has spurred the development of novel agents for the treatment of AML, including new oral targeted therapies (midostaurin, gilteritinib, enasidenib, and ivosidenib) that have recently received regulatory approval.
Acute myeloid leukemia (AML), also known as acute myelogenous leukemia or acute nonlymphocytic leukemia, is a heterogeneous disease in terms of its underlying genetics, pathobiology, and clinical manifestations.1,2 This hematologic malignancy is characterized by differentiation arrest and uncontrolled proliferation of precursor cells (blasts) of the myeloid lineage—i.e., progenitors that give rise to granulocytic, monocytic, erythroid, or megakaryocytic cells, resulting in impaired hematopoiesis and/or bone marrow failure—within the hematopoietic system and infiltration of tumors to other tissues (e.g., extramedullary disease).1 As a result, the reduction in normal granulocytes, erythrocytes, and platelets is variable, leading to systemic consequences including anemia, bleeding, and increased risk of infection. If AML is left untreated, patients succumb to coagulopathy and infections within months after initial diagnosis.
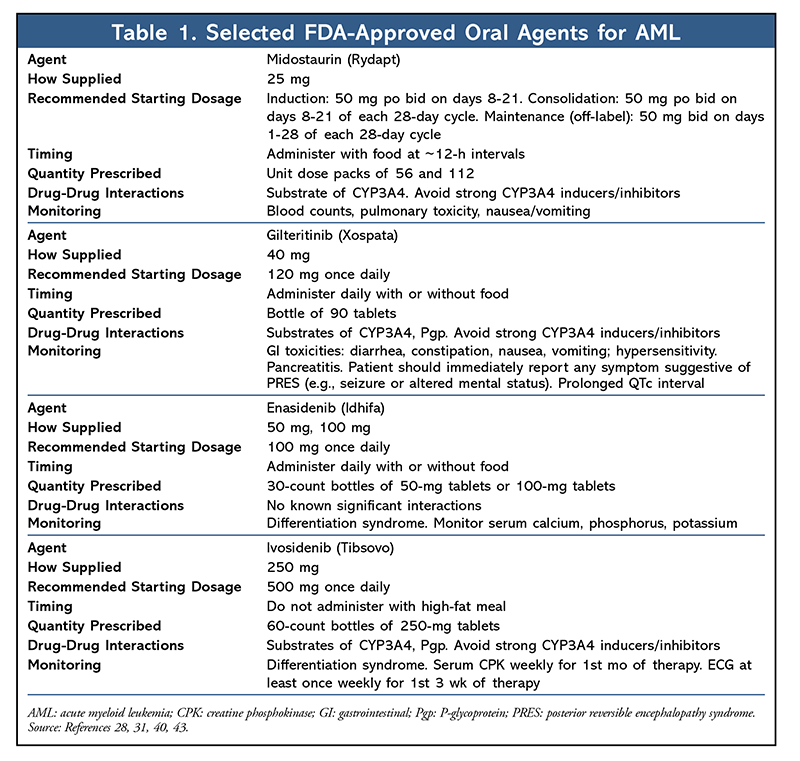
AML, the most common form of leukemia in adults, accounts for the largest number of annual deaths from leukemia in the United States.1 Although many AML patients initially achieve remission after induction chemotherapy, relapse and refractory disease are the major causes of treatment failure, which underscores the unmet need for novel therapies.2 Prior to 2017, acute promyelocytic leukemia (APL) was the only type of AML for which targeted therapy (all-trans-retinoic acid) was mandatory in routine practice.3 Recently, much excitement has been generated by the emergence and FDA approval of several oral targeted therapies for AML (TABLE 1). Each of these therapies has unique pharmacology, dosing, and adverse effects, necessitating close monitoring to mitigate adverse effects, improve compliance, and ensure therapeutic benefit. It is important for pharmacists to be cognizant of the recent advances in AML and be able to apply this knowledge in clinical practice.
Diagnosis and Disease Biology
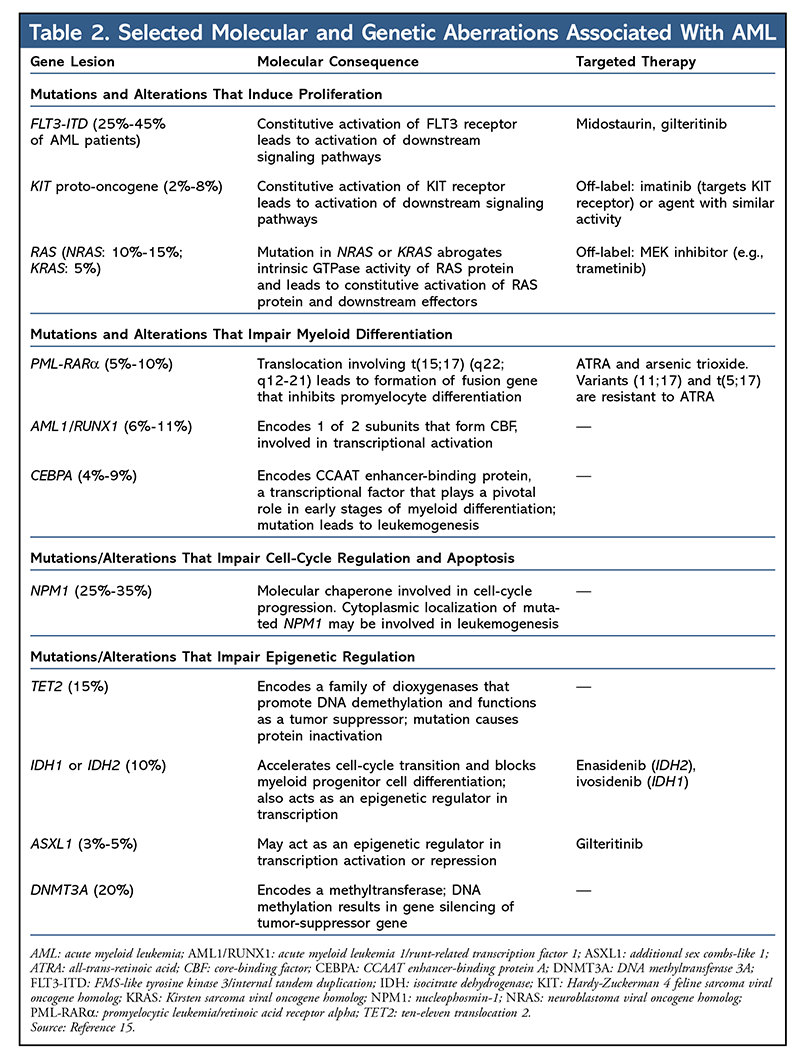
A diagnosis of AML requires morphological assessment of peripheral blood and bone marrow, analysis of the expression of cell-surface or cytoplasmic markers through flow cytometry (i.e., immunophenotyping), identification of chromosomes by cytogenetics, and screening for selected molecular or genetic lesions that are used to classify different subtypes of AML (TABLE 2).1
Historically, AML has been classified by the largely descriptive French-American-British criteria, which are based on morphology and cytochemical stains.4 In the last two decades, the discovery of novel genomic and molecular biomarkers has changed the paradigms for disease prognosis and treatment decisions. The World Health Organization (WHO) included genomic and molecular data in the 2016 revision of its AML classification. The 2016 classification aims to identify biological entities (or subsets of patients) that could elucidate molecular or signaling pathways that are amenable to targeted therapies. The major categories of the 2016 classification include AML with recurrent genetic abnormalities, AML with myelodysplasia-related changes, therapy-related AML, and AML not otherwise specified.5 The 2016 classification specifies that AML be diagnosed based on the presence of ≥20% of blasts in the bone marrow or peripheral blood; notably, however, the presence of clonal structural cytogenetic abnormalities—e.g., t(15;17), t(8;21), inv(16), or t(16;16)—permits a diagnosis of AML regardless of the percentage of blasts.
Molecular Pathogenesis
AML is a clonal disease characterized by the presence of a variety of genetic alterations. Most cases of AML are distinguished by clonal heterogeneity at the time of diagnosis, with the presence of a founding clone and at least one subclone.6 Various patterns of clonal evolution (presence of additional genetic abnormalities in leukemic blasts) at relapse probably contribute to resistance to therapy.7 Studies using cytogenetic analysis showed that recurrent chromosomal structural variations (karyotype) are characterized by acquired genetic abnormalities (i.e., somatic mutations) that have essential roles in the pathogenesis of leukemia (leukemogenesis).8,9 The karyotype of the leukemic cells represents the most important prognostic factor for AML in the last two decades.
Recently, the discovery of molecular and/or genetic alterations has led to the refinement of prognostication in AML.5,6,10-12 Targeted DNA sequencing has identified recurrent mutations in FLT3, NPM1, KIT, CEBPA, and TET2 (see TABLE 2 for an annotated summary of these genes and their molecular consequences).10-12 These genes normally participate in normal myeloid cell differentiation and self-renewal. When these genes are altered, they may contribute to leukemogenesis. Genes that are commonly involved in epigenetic regulation of myeloid cell differentiation (e.g., DNMT3A, ASXL1, IDH2, and TET2) are also present in preleukemic hematopoietic stem cells and occur early in the evolution of AML.13,14 These preleukemic stem cells are capable of multilineage differentiation and can survive chemotherapy, eventually leading to relapse.
Risk Stratification and Treatment Options
AML treatment relies on eradication of the leukemic blasts through remission-induction chemotherapy (typically a combination of an anthracycline and cytarabine) in selected patients to induce morphological remission, followed by consolidation (postremission) chemotherapy to maintain disease remission and restore normal hematopoiesis in the bone marrow.15 Unfortunately, owing to the heterogeneous cytogenetic and molecular abnormalities of leukemic blasts, a significant number of patients eventually experience disease relapse.1 Patients with relapsed or refractory disease often have a poor prognosis and limited treatment options. Improved understanding of the disease pathogenesis has led to new therapeutic approaches.
In general, patients whose cytogenetic profile is associated with a favorable risk have relatively good outcomes from remission-induction chemotherapy and chemotherapy-based consolidation regimens and therefore do not require allogeneic hematopoietic stem-cell transplantation (HSCT), whereas patients with an unfavorable-risk (poor-risk) profile (e.g., those with complex cytogenetics) require allogeneic HSCT during the first complete remission in order to improve prognosis.16,17 Subsequently relapsed patients, regardless of their cytogenetics, are classified as being poor-risk. However, most AML patients have an intermediate cytogenetic risk, which is characterized by a normal karyotype. Some of these patients do well with chemotherapeutic consolidation, whereas others have an extremely poor outcome; for this reason, recent studies have incorporated new biomarkers for better classification of intermediate risk.12,18 Newer classification algorithms incorporate FLT3, NPM1, CEBPA, and KIT into standard-of-care testing. Recent studies have revealed that mutations in newly discovered AML genes (e.g., DNMT3A, IDH1/2, and TET2) provide prognostic information for patients with an intermediate-risk profile.12,18,19
New Therapies
The frequent occurrence of mutations or gene alterations has led to the development of novel agents for the treatment of AML. These agents target a variety of cellular processes, such as signaling pathways in the differentiation of myeloid precursor cells, genes involved in DNA methylation and chromatin remodeling, and antigens on leukemic cells.20 Some novel oral targeted therapies that recently received regulatory approval are discussed below.
FLT3 Inhibitors: The FLT3 gene encodes a receptor tyrosine kinase involved in hematopoiesis. The FLT3 receptor is expressed by hematopoietic stem cells to control stem-cell replication, differentiation, and persistence.21 FLT3 mutations are not a leukemic initiating event, but rather a “late hit” that may account for disease progression.22 Other late-hit targets may include gene alterations in NRAS and IDH1.13 In fact, FLT3 is one of the most frequently mutated genes in AML, with mutations occurring in up to 30% of patients.23,24 There are two distinct activating FLT3 mutations: internal tandem duplications (ITD) in the juxtamembrane domain of the transmembrane protein and point mutations in the tyrosine kinase domain (TKD), most commonly at codon aspartic acid 835 (D835).25,26 FLT3-ITD or FLT3-TKD mutation leads to constitutive activation of receptor tyrosine kinase, which contributes to leukemogenesis. FLT3-ITD mutation is associated with an aggressive disease course and is a prognostic marker for rapid relapse and short overall survival after chemotherapy.24 FLT3 inhibitors used as single agents often lead to transient reductions in blast counts.27
Midostaurin: April 28, 2017, marked the first FDA approval of a targeted therapy for AML since a targeted therapy for APL was approved; approval was based on the results of a phase III randomized trial (RATIFY, also known as CALGB 10603) in FLT3 mutation–positive newly diagnosed AML patients.28 In this trial of patients who received midostaurin (Rydapt), a multitargeted kinase inhibitor, or placebo in combination with standard induction and consolidation chemotherapy, midostaurin patients had improved overall survival (hazard ratio 0.77; 95% CI, 0.63-0.95; P = .007), although the rate of grade ≥3 anemia was higher in the midostaurin group than in the placebo group (92.7% vs. 87.8%; P = .03), as was the rate of grade ≥3 rash (14.1% vs. 7.6%; P = .008).
There are several takeaways from RATIFY: 1) It took about 10 years to complete this trial for a rare disease that requires rapid treatment, and successful enrollment required the ability to conduct rapid genetic testing. 2) The low rate of FLT3-ITD mutations was due to the higher-than-anticipated rate of HSCT and disproportionate number of patients with FLT3-TKD mutations. However, the benefit of midostaurin is not abrogated by HSCT in first complete remission. 3) The benefit of midostaurin did not vary by type of FLT3 mutation (ITD vs. TKD) or ratio of mutant to wild-type ITD allele (high vs. low), suggesting that some newly diagnosed patients may not be highly dependent on FLT3 signaling; further studies are therefore warranted. 4) A significant number of HSCT patients (61 of 128) were able to initiate midostaurin maintenance.28 However, FLT3 inhibitors used as maintenance therapy for AML have not received regulatory approval, and their role is still being investigated. As is the case with other investigational FLT3 inhibitors, studies examining the role of midostaurin in relapsed or refractory AML are ongoing.
Gilteritinib: In 2018, gilteritinib (Xospata), an oral FLT3 and AXL inhibitor designed to target FLT3-ITD mutations and address the limitations of midostaurin (e.g., brief clinical response, lack of mutation selectivity), received regulatory approval based on an interim analysis of the ADMIRAL trial (NCT02421939).29 This trial included 138 adult patients with relapsed or refractory AML with mutations in FLT3-ITD or FLT3-TKD, such as D835 or I836 mutations.30 Gilteritinib 120 mg daily was given orally until unacceptable toxicity or lack of clinical benefit. After a median follow-up of 4.6 months (range, 2.8-15.8 months), 29 patients achieved complete remission (CR) or CR with partial hematologic recovery (21%; 95% CI, 14.5-28.8).
The most common adverse reactions (occurring in ≥20% of patients) included myalgia/arthralgia, transaminase increase, fatigue/malaise, noninfectious diarrhea, and rash, among others. QTc-interval prolongation occurred in about 1.4% of gilteritinib-treated patients.31 The manufacturer recommends a baseline ECG prior to treatment with gilteritinib, on days 8 and 15 of cycle 1 and prior to the start of the subsequent two cycles. Dose interruption or reduction is warranted in patients who have a QTc >500 msec.31 As expected in patients with relapsed or refractory AML, neutropenic infections were common (>50%), but treatment-related mortality from neutropenia was generally <1%.32
The benefit of combining chemotherapy with small-molecule tyrosine kinase inhibitors may not apply to all broad-spectrum FLT3 kinase inhibitors. One trial showed that sorafenib used as a single agent after allogeneic HSCT could enhance treatment response in patients with FLT3-ITD mutations.33 Interestingly, FLT3-mutated patients achieved a CR with incomplete hematologic recovery, which suggests that minimal residual disease persisted after gilteritinib treatment. This can be explained by the fact that administration of a single FLT3 inhibitor to a genetically complex bone-marrow environment may unmask the underlying ineffective hematopoiesis capacity of the remaining bone-marrow progenitor cells, causing erythropoiesis and megakaryopoiesis to be persistently suppressed by the FL3 inhibitor.32
IDH Inhibitors: Isocitrate dehydrogenase (IDH) comprises a family of enzymes involved in the cellular defense of oxidative damage.33 IDH1 is localized in cytoplasm, whereas IDH2 is found in mitochondria. IDH1 mutation, which was identified by sequencing an entire AML genome, was strongly associated with cytogenetically normal AML. IDH mutations in human malignancies exclusively affect codon arginine 132 (R132) and codon arginine 172 (R172) in IDH1 and IDH2 (more frequent in AML), respectively.34,35 Patients with IDH mutations were older, had lower WBC counts, and exhibited higher platelet counts.36 IDH1 or IDH2 mutations tend to be mutually exclusive and confer a neomorphic enzymatic activity, resulting in the reduction of alpha-ketoglutarate to the oncometabolite 2-hydroxyglutarate and leading to epigenetic alterations and impaired hematopoietic differentiation.37,38
Enasidenib: Enasidenib (Idhifa) is an IDH2 inhibitor. This agent received regulatory approval in 2017 based on a 26.6% CR plus CR with partial hematologic response in a phase I/II trial of IDH2-mutant patients with relapsed or refractory AML at 100 mg orally daily.39 An additional 12% of patients either experienced partial response or achieved a morphological leukemia-free state. Patients with clinical response did not experience a reduction in IDH2-mutant allele burden, reflecting a shift from undifferentiated to differentiated clonal hematopoiesis. The most common adverse effects from enasidenib were nausea and indirect hyperbilirubinemia. The latter is thought to be caused by the off-target inhibition of UGT1A1 enzyme in the liver and is not considered clinically significant; therefore, dose reduction is not required, and hyperbilirubinemia generally resolves with continuous treatment.39
In the trial, differentiation syndrome (DS) was seen in about 14% of patients, and onset occurred as early as 10 days and up to the first 5 months after treatment initiation.40 As seen in therapy for APL, DS is due to a proliferation of differentiated leukemic cells that alters cytokine balance, leading to tissue damage and inflammation. Signs and symptoms of DS include unexplained fever, weight gain, respiratory symptoms, third spacing, and acute kidney injury. If symptoms are severe, the manufacturer and investigators recommend starting a corticosteroid immediately and withholding enasidenib until symptoms resolve or improve to grade ≤2.40,41
Ivosidenib: Ivosidenib (Tibsovo), an IDH1 inhibitor, received regulatory approval based on an open-label, single-arm, multicenter trial (NCT02074839) demonstrating a CR plus CR with a partial hematologic recovery rate of 32.8% (95% CI, 25.8%-40.3%).42 Median time to response (range, 0.9-5.6 months) was 2 months, and median response duration was 8.2 months (95% CI, 5.6-12 months). Of the 110 patients who were transfusion-dependent at baseline, 41 (37.3%) became independent of transfusions during any 56-day postbaseline period. The most common adverse reactions (≥20%) were fatigue, leukocytosis, arthralgia, diarrhea, dyspnea, edema, nausea, mucositis, prolonged QT on ECG, rash, pyrexia, cough, and constipation. The reported rate of DS in the trial was 19% (34 of 179 patients).43
Conclusion
FDA approvals of new oral targeted therapies for AML are the result of several decades of research that brought improved diagnostic and therapeutic strategies to the clinic. The development of these agents has fostered the hope that additional therapies targeting different molecular abnormalities in AML may be forthcoming. Ultimately, as researchers gain insight into the genetic and epigenetic changes relevant to the pathogenesis of AML, the identification of novel molecular and genetic markers will contribute to an understanding of leukemia’s biology, leading to improvements in patient risk assessment and the eventual development of novel therapies targeting these molecular or genetic changes.
REFERENCES
1. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines). Acute myeloid leukemia. Version 1.2019. www.nccn.org. Accessed January 26, 2019.
2. Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136-1152.
3. Lo-Coco F, Cicconi L, Breccia M. Current standard treatment of adult acute promyelocytic leukaemia. Br J Haematol. 2016;172(6):841-854.
4. Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol. 1976;33(4):451-458.
5. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405.
6. Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059-2074.
7. Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506-510.
8. Rowley JD. Chromosomal translocations: revisited yet again. Blood. 2008;112(6):2183-2189.
9. Mrózek K, Heerema NA, Bloomfield CD. Cytogenetics in acute leukemia. Blood Rev. 2004;18(2):115-136.
10. Stirewalt DL, Radich JP. The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer. 2003;3(9):650-665.
11. Bacher U, Schnittger S, Haferlach T. Molecular genetics in acute myeloid leukemia. Curr Opin Oncol. 2010;22(6):646-655.
12. Patel JP, Gönen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079-1089.
13. Krönke J, Bullinger L, Teleanu V, et al. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood. 2013;122(1):100-108.
14. Corces-Zimmerman MR, Hong WJ, Weissman IL, et al. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci U S A. 2014;111(7):2548-2553.
15. Chung C, Ma H. Driving toward precision medicine for acute leukemias: are we there yet? Pharmacotherapy. 2017;37(9):1052-1072.
16. Breems DA, Van Putten WL, De Greef GE, et al. Monosomal karyotype in acute myeloid leukemia: a better indicator of poor prognosis than a complex karyotype. J Clin Oncol. 2008;26(29):4791-4797.
17. Byrd JC, Mrózek K, Dodge RK, et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461). Blood. 2002;100(13):4325-4336.
18. Mrózek K, Marcucci G, Nicolet D, et al. Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. J Clin Oncol. 2012;30(36):4515-4523.
19. Shen Y, Zhu YM, Fan X, et al. Gene mutation patterns and their prognostic impact in a cohort of 1185 patients with acute myeloid leukemia. Blood. 2011;118(20):5593-5603.
20. Kayser S, Levis MJ. Advances in targeted therapy for acute myeloid leukaemia. Br J Haematol. 2018;180(4):484-500.
21. Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100(5):1532-1542.
22. Shih LY, Huang CF, Wang PN, et al. Acquisition of FLT3 or N-ras mutations is frequently associated with progression of myelodysplastic syndrome to acute myeloid leukemia. Leukemia. 2004;18(3):466-475.
23. The Cancer Genome Atlas Research Network, Ley TJ, Miller C, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059-2074.
24. Thiede C, Steudel C, Mohr B, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99(12):4326-4335.
25. Nakao M, Yokota S, Iwai T, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10(12):1911-1918.
26. Yamamoto Y, Kiyoi H, Nakano Y, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97(8):2434-2439.
27. Wander SA, Levis MJ, Fathi AT. The evolving role of FLT3 inhibitors in acute myeloid leukemia: quizartinib and beyond. Ther Adv Hematol. 2014;5(3):65-77.
28. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454-464.
29. Fischer T, Stone RM, Deangelo DJ, et al. Phase IIB trial of oral midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol. 2010;28(28):4339-4345.
30. Gorcea CM, Burthem J, Tholouli E. ASP2215 in the treatment of relapsed/refractory acute myeloid leukemia with FLT3 mutation: background and design of the ADMIRAL trial. Future Oncol. 2018;14(20):1995-2004.
31. Xospata (gilteritinib) package insert. Northbrook, IL: Astellas Pharma Inc; November 2018.
32. Perl AE, Altman JK, Cortes J, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1-2 study. Lancet Oncol. 2017;18(8):1061-1075.
33. Metzelder SK, Schroeder T, Finck A, et al. High activity of sorafenib in FLT3-ITD-positive acute myeloid leukemia synergizes with allo-immune effects to induce sustained responses. Leukemia. 2012;26(11):2353-2359.
34. Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361(11):1058-1066.
35. Park SW, Chung NG, Han JY, et al. Absence of IDH2 codon 172 mutation in common human cancers. Int J Cancer. 2009;125(10):2485-2486.
36. Paschka P, Schlenk RF, Gaidzik VI, et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol. 2010;28(22):3636-3643.
37. Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alphaketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17(3):225-234.
38. Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553-567.
39. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722-731.
40. Idhifa (enasidenib) package insert. Summit, NJ: Celgene Corp; August 2018.
41. Fathi AT, DiNardo CD, Kline I, et al. Differentiation syndrome associated with enasidenib, a selective inhibitor of mutant isocitrate dehydrogenase 2: analysis of a phase 1/2 study. JAMA Oncol. 2018;4(8):1106-1110.
42. DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378(25):2386-2398.
43. Tibsovo (ivosidenib) package insert. Cambridge, MA: Agios Pharmaceuticals, Inc; July 2018.
To comment on this article, contact rdavidson@uspharmacist.com.



