US Pharm. 2020:45; (7)(Specialty& Oncology suppl):8-12.
ABSTRACT: Sickle cell disease is a group of inherited blood disorders in which patients are born with sickled hemoglobin. As a result, patients are at an increased risk for complications associated with anemia and vaso-occlusion. Hydroxyurea and blood transfusions have been the gold standard of therapy for the management of sickle cell disease. Despite current therapies, sickle cell patients still experience frequent anemia and vaso-occlusive crises yearly. Recently, three novel agents were approved to reduce the incidence of vaso-occlusive pain crises and/or anemia in sickle cell patients. Current treatment options and these novel agents can be used to successfully manage these two major complications of sickle cell disease.
Sickle cell disease is a group of inherited red blood cell disorders in which patients inherit two defective hemoglobin genes. Sickle cell disease is the most common genetic disease in the United States and affects approximately 100,000 Americans.1 Sickle cell disease occurs predominantly in African American and Hispanic American patients; one in every 365 African American and one in every 16,300 Hispanic American children are born with sickle cell disease.2 Sickle cell disease also has a global impact, with a high prevalence among people of sub-Saharan Africa, South America, the Caribbean, Central America, Saudi Arabia, India, and the Mediterranean.
Sickle cell disease is caused by a mutation in which glutamine is replaced with valine at the sixth position of the beta globin gene (HBB) and is characterized by the production of sickled hemoglobin (HgS). Sickle cell disease encompasses a group of disorders in which at least one HgS allele is present. Sickle cell anemia, HgSS, is the most common disorder; it occurs when a patient inherits both HgS alleles and accounts for 60% to 70% of patients with sickle cell disease.3 Other common disorders include sickle–hemoglobin C disease and sickle beta thalassemia, both of which are caused when a patient inherits one HgS allele and another abnormal beta-globin chain mutation.
PATHOGENESIS
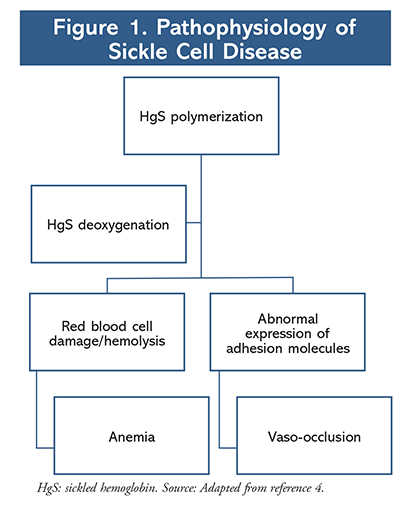
Polymerization is a key pathway in the pathogenesis of sickle cell disease (FIGURE 1). Upon deoxygenation of hemoglobin, HgS undergoes polymerization, which damages the blood cell.4 As a result, a more rigid, adhesive HgS is produced that sticks to other blood cells and endothelium and in turn occludes small blood vessels. The occlusion of small blood vessels can lead to tissue ischemia which may present as vaso-occlusive pain crisis, acute chest syndrome, stroke, or organ failure. Polymerization also reduces the life span of HgS and therefore leads to hemolysis and anemia in sickle cell patients. Vaso-occlusive pain crises and anemia are hallmark complications associated with sickle cell disease.
PREVENTION
Hydroxyurea and blood transfusions have been the mainstays in the prevention of anemia and vaso-occlusive crises in sickle cell disease. Hydroxyurea is an antimetabolite proven to reduce the incidence of vaso-occlusive pain crises and the need for blood transfusions in sickle cell patients.5 Despite the efficacy of current therapeutic options, sickle cell patients experience an average of three pain crises yearly, and 96% of hospitalizations of sickle cell patients are due to vaso-occlusive crisis.6,7 Sickle cell patients also have a 33%, 30-day hospital-readmission rate; most readmissions are usually related to vaso-occlusive crises.7 In 2018, the annual healthcare expenditure for sickle cell–related hospitalizations was estimated at over $900 million.8
Hematopoietic stem cell transplantation (HSCT) is currently the only curative therapy for patients with sickle cell disease. HSCT was found to have an 87% curative rate among sickle cell patients with severe disease.9 Unfortunately, HSCT is an option for only a small number of patients, typically pediatric patients with severe sickle cell disease; use is rare in adults. Several factors such high cost, severe side effects post-transplantation, risk of death, and the requirement for an exact donor match limit use among sickle cell patients.10 There has been a desperate need for additional therapies to increase life expectancy and to reduce complications associated with sickle cell disease. The average life expectancy of a sickle cell disease patient is approximately 2 to 3 decades less than that of an American without sickle cell disease.11,12
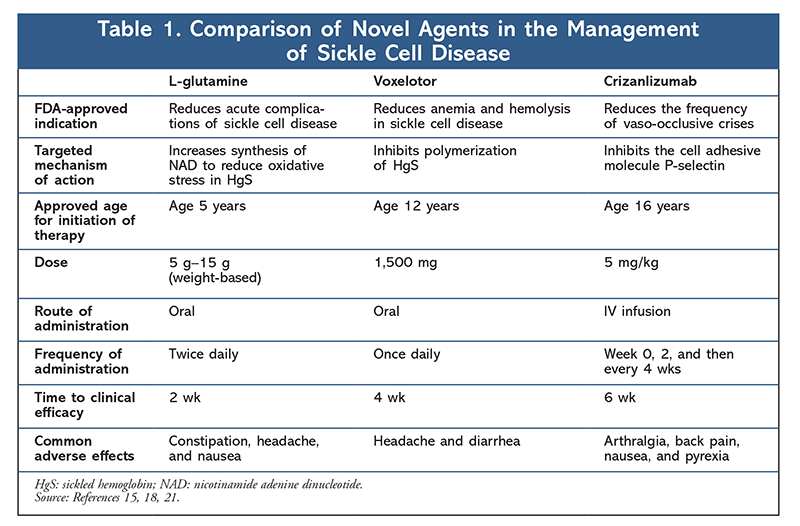
Recently, three novel agents (TABLE 1) have been approved as an advancement to current therapies to reduce the incidence of vaso-occlusive crises and the risk of anemia among sickle cell patients. This article will focus on the two major complications associated with sickle cell disease and the current treatment options used to manage them.
ANEMIA
Sickle cell patients are naturally predisposed to anemia secondary to the shortened half-life of HgS. Typically, red blood cells have an average half-life of 120 days; however, HgS has a half-life of 10 to 20 days.13 Most patients with sickle cell disease have chronic anemia, and baseline hemoglobin levels may vary according to hemoglobin genotype. Acute anemia in sickle cell disease is defined as a reduction in the hemoglobin concentration by 2 g/dL.14 Blood transfusions, a cornerstone of treatment for anemia, improve anemia by increasing the concentration of normal hemoglobin (HgA) to reduce the concentration of HgS. Blood transfusions are indicated in patients who have symptomatic anemia or who experience complications such as acute splenic sequestration, an aplastic crisis, acute chest syndrome, or infection, which can all worsen anemia. Although effective and lifesaving, blood transfusions come with risks such as alloimmunization, infection, and iron overload, which can limit use.
Other than blood transfusions, until recently there have been no therapeutic options indicated to prevent or reduce the incidence of anemia associated with sickle cell disease. In 2019, Oxbryta (voxelotor) was approved for the treatment of sickle cell disease in patients aged 12 years or older. Voxelotor is a HgS polymerization inhibitor that blocks the initial step in the production of HgS and works to stabilize oxygenation to reduce sickling of hemoglobin.15
Voxelotor
The HOPE trial evaluated the use of voxelotor versus placebo in sickle cell patients who had a hemoglobin level between 5.5 and 10.5 g/dL and who had experienced one to 10 pain crises within a year.16 The primary endpoint, an increase in hemoglobin by at least 1 g/dL at week 24, was achieved in 51% of the patients who received voxelotor compared with 7% of the patients who received placebo, regardless of hydroxyurea use (P <.001). Voxelotor also significantly reduced indirect bilirubin (-29.08% vs. 3.16%; P <.001) and the percentage of reticulocytes (-19.93% vs. 4.54%; P <.001), which is indicative of its ability to reduce anemia and hemolysis. Voxelotor was also shown to have a greater reduction in the annual incidence of vaso-occlusive pain crisis when compared with placebo. There were no thrombotic events reported during the 24-week trial.
Voxelotor is commercially available as an oral formulation that is dosed once daily with or without regard to food. Dose adjustments are required in patients with severe hepatic impairment. There are no contraindications with use; however, patients should be monitored for hypersensitivity reactions. Voxelotor should not be used in pregnant women or in women who are breastfeeding. The most common adverse effects include headache, diarrhea, abdominal pain, nausea, fatigue, rash, and pyrexia.
VASO-OCCLUSIVE CRISES
Vaso-occlusive crisis is a complication of sickle cell disease that results from occlusion of HgS in the microvasculature. Almost all sickle cell patients will experience at least one vaso-occlusive crisis within their lifetime.14 Patients with vaso-occlusive crises present with sudden, excruciating pain, commonly in the back, chest, and/or extremities. Acute treatment of vaso-occlusive crisis may include hydration, pain management, and blood transfusions. Blood transfusions are also used chronically to prophylactically to treat patients who are at an increased risk for recurrent vaso-occlusive events. Since 1998, hydroxyurea has been used first-line to prevent the occurrence of vaso-occlusive pain crises in sickle cell patients. Although efficacious, hydroxyurea is associated with hematologic and teratogenic toxicities which require frequent monitoring. Hydroxyurea also has an unfavorable side-effect profile, a high rate of nonadherence, and is often underprescribed by healthcare professionals.17
L-Glutamine
In 2017, Endari (L-glutamine) was approved to reduce the frequency of pain crises in sickle cell patients older than age 5 years who experience two or more pain crises a year. L-glutamine is an essential amino acid that reduces oxidative stress in sickled red blood cells by synthesizing nicotinamide adenine dinucleotide (NAD).18 NAD plays a key role in preventing cell damage in red blood cells, which reduces sickling of hemoglobin and prevents adhesion of red blood cells to endothelium.19
L-glutamine was studied in sickle cell patients with or without concomitant use of hydroxyurea in a multicenter, randomized, double-blinded, placebo-controlled, phase III, 48-week study.20 L-glutamine was shown to reduce the frequency of vaso-occlusive pain crises by 25% (3.2 vs. 3.9 events per year; P = .005), and only 8.6% of the patients who received L-glutamine experienced at least one pain crisis within the year following use. L-glutamine was also shown to prolong the duration between the first and second occurrence of pain crises and to reduce the frequency of hospitalizations by 33% (2 vs. 3 mean annual hospitalizations; P = .005). Unfortunately, only 63% of patients who received L-glutamine completed the study protocol; however, only 3.3% of patients discontinued use due to adverse effects.
L-glutamine is commercially available as an oral-powder packet that must be dissolved in 8 ounces of a beverage or eaten with 4 to 6 ounces of food such as applesauce or yogurt.18 L-glutamine dosing is weight-based and is dosed twice daily. There are no required dose adjustments with use. There are also no reported contraindications with use; however, caution should be used in patients with renal and hepatic impairment. L-glutamine should not be used in pregnant women or in women who are breastfeeding. The most common adverse effects include constipation, headache, and nausea.
Crizanlizumab
The most notable addition to therapies indicated for the prevention of vaso-occlusive crises is Adakveo (crizanlizumab). Crizanlizumab is a monoclonal antibody that inhibits P-selectin.21 Vaso-occlusion is caused by the expression of P-selectin, which results in the adhesion of HgS and leukocytes to endothelium. Crizanlizumab was approved in late 2019 to reduce the incidence of vaso-occlusive pain crises in sickle cell patients older than age 16 years who experience frequent pain crises.
In the SUSTAIN trial, crizanlizumab was effective in reducing the incidence of pain crises by 45.3% (P = .01) when compared with placebo, regardless of hydroxyurea use.22 Better outcomes were observed in patients who were not using hydroxyurea at baseline. More than a third of the patients who received crizanlizumab did not experience a vaso-occlusive crisis, and patients with a reported incidence of five to 10 pain crises at baseline experienced a 63% reduction. Crizanlizumab was also shown to prolong the duration between the first and second occurrence of pain crisis, and it reduced the annual rate of days hospitalized by 41.8%.
Crizanlizumab is the first parenteral agent approved for the management of vaso-occlusive crises. It is a once-monthly IV infusion and requires no dose adjustments with use. There are no reported contraindications with use; however, patients should be monitored for severe infusion-related reactions. Immunogenicity may also occur with use. Crizanlizumab should not be used in pregnant women or in women who are breastfeeding. The most common adverse effects with use are nausea, arthralgia, back pain, and pyrexia.
Future Advances in Therapy
Gene therapy is currently being studied to provide another curative option in sickle cell patients. Gene therapy uses genetic material from patients to cure their own disease. Similar to HSCT, patients still receive chemotherapy to eliminate or reduce defective blood stem cells. However, gene therapy uses a lentiviral vector, which is a specialized biological agent, to insert corrected genes back into cells to repair or replace the patient’s genes.23 The novel gene transferred back into the patient is thought to increase the amount of fetal hemoglobin and reduce the amount of HgS to cure sickle cell disease. An open-label, nonrandomized, single-center study is active and is currently in phase I. The study is scheduled to complete in early 2024.
In April 2020, FT-4202 was designated as an orphan drug for the treatment of sickle cell anemia. FT-4202 is a pyruvate kinase-R (PKR) activator that assists in maintaining the integrity and oxygenation of red blood cells to prevent sickling.24 If successful, FT-4202 is proposed to be a disease-modifying agent that will inhibit a pivotal initial step in the cascade of events that leads to complications associated with sickle cell disease. It is presumed to effectively reduce the incidence of vaso-occlusive pain crises and anemia among sickle cell patients. A randomized, double-blinded, placebo-controlled phase I study is currently active for healthy and sickle cell patients.25
CONCLUSION
Sickle cell patients with frequent hospitalizations related to vaso-occlusive crisis are at an increased risk for early death. For more than two decades, hydroxyurea has been the gold standard in preventing the incidence of vaso-occlusive crises and anemia and reducing the need for blood transfusion in sickle cell patients. Unfortunately, sickle cell patients still experience frequent vaso-occlusive pain crises and chronic anemia despite current therapy.
Crizanlizumab, L-glutamine, and voxelotor reduced the incidence of vaso-occlusive events, and voxelotor was also beneficial in reducing anemia and hemolysis in sickle cell patients. New therapies, specifically L-glutamine and voxelotor, may be used as adjunct therapy to hydroxyurea or in place of hydroxyurea if intolerable. Due to its profound results, crizanlizumab may at some point compete as first-line therapy in place of hydroxyurea in a specific population of patients, with continued data on its long-term effects. As a result of new targeted therapies, there are now more therapeutic options that can be used to reduce the incidence of anemia and sickle cell–related pain crises. With potential investigational targeted and curative therapies on the horizon, more advances in mitigating complications or completely eradicating sickle cell disease can be anticipated in the future.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
REFERENCES
1. Sedrak A, Kondamudi NP. Sickle cell disease. Updated December 13, 2019. In: StatPearls [Internet]. Treasure Island, FL: StatPearls Publishing; January 2020. www.ncbi.nlm.nih.gov/books/NBK482384/. Accessed July 15, 2020.
2. CDC. Data and statistics on sickle cell disease. www.cdc.gov/ncbddd/sicklecell/data.html. Accessed April 4, 2020.
3. Bender MA. Sickle cell disease. 2003 September 15, 2003; updated August 17, 2017. In: Adam MP, Ardinger HH, Pagon RA, et al, eds, GeneReviews [Internet]. Seattle, WA: University of Washington, Seattle; 1993-2020. www.ncbi.nlm.nih.gov/books/NBK1377/. Accessed July 15, 2020.
4. Eaton W, Hofrichter J. Sickle cell hemoglobin polymerization. Adv in Protein Chem. 1990;40:63-279.
5. Droxia (hydroxyurea) prescribing information. Princeton, New Jersey: Bristol-Myers Squibb Company; 2019.
6. Neumayr LD, Hoppe CC, Brown C. Sickle cell disease: current treatment and emerging therapies. Am J Manag Care. 2019;25(18 Supp0:S335-S343.
7. Fingar KR, Owens PL, Reid LD, et al. Statistical Brief #251. Healthcare Cost and Utilization Project (HCUP). Characteristics of inpatient hospital stays involving sickle cell disease, 2000-2016. September 2019. Agency for Healthcare Research and Quality, Rockville, MD. www.hcup-us.ahrq.gov/reports/statbriefs/sb251-Sickle-Cell-Disease-Stays-2016.jsp. Accessed May 5, 2020.
8. Bou-Maroun LM, Meta F, Hanba CJ, et al. An analysis of inpatient pediatric sickle cell disease: incidence, costs, and outcomes. Pediatr Blood Cancer. 2018;65(1):e26758.
9. Hsieh M, Fitzhugh C, Weitzel R, et al. Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA. 2014;312(1):48-56.
10. Shenoy S. Hematopoietic stem-cell transplantation for sickle cell disease: current evidence and opinions. Ther Adv Hematol. 2013;4(5):335-344.
11. Piel FB, Steinberg MH, Rees DC. Sickle cell disease. N Engl J Med. 2017;376(16):1561-1573.
12. Lubeck D, Agodoa I, Bhakta N, et al. Estimated life expectancy and income of patients with sickle cell disease compared with those without sickle cell disease. JAMA Netw Open. 2019;2(11):e1915374.
13. Medline Plus. Sickle cell disease. Sickle cell anemia. https://medlineplus.gov/sicklecelldisease.html. Accessed May 5, 2020.
14. National Heart, Lung, and Blood Institute. U.S. Department of Health and Human Services. Evidence-based management of sickle cell disease: Expert Panel Report 2014. www.nhlbi.nih.gov/health-topics/evidence-based-management-sickle-cell-disease. Accessed July 15, 2020.
15. Oxbryta (voxelotor) prescribing information. South San Francisco, CA: Therapeutics, Inc; 2019.
16. Vichinsky E, Hoppe C, Atoga K, et al. A phase 3 randomized trial of voxelotor in sickle cell disease. N Engl J Med. 2019;381(6):509-519.
17. Shah N, Bhor M, Xie L, et al. Treatment patterns and economic burden of sickle-cell disease patients prescribed hydroxyurea: a retrospective claims-based study. Health Qual Life Outcomes. 2019;17(1):155.
18. Endari (L-glutamine) prescribing information. Torrance, CA: Emmaus Medical, Inc; 2017.
19. Niihara Y, Zerez CR, Akiyama DS, Tanaka KR. Increased red cell glutamine availability in sickle cell anemia: demonstration of increased active transport, affinity, and increased glutamate level in intact red cells. J Lab Clin Med. 1997;130:83-90.
20. Niihara Y, Miller ST, Kanter J, et al. A phase 3 trial of L-glutamine in sickle cell disease. N Engl J Med. 2018;379(3):226-235.
21. Adakveo (crizanlizumab) prescribing information. East Hanover, NJ: Novartis; 2019.
22. Ataga K, Kutlar A, Kanter JI, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. 2017;365(5):429-439.
23. ClinicalTrials.gov. Gene transfer for sickle cell disease. https://clinicaltrials.gov/ct2/show/study/NCT03282656. Accessed May 11, 2020.
24. FT-4202. Sickle cell disease. www.formatherapeutics.com/pipeline/ft-4202/. Accessed May 11, 2020.
25. ClnicalTrials.gov. A SAD/MAD to assess the safety, pharmacokinetics and pharmacodynamics of FT-4202 in healthy volunteers and sickle cell disease patients. https://clinicaltrials.gov/ct2/show/NCT03815695?term=%22FORMA+Therapeutics%22&rank=3. Accessed May 11, 2020.
To comment on this article, contact rdavidson@uspharmacist.com.



