US Pharm. 2020;45(9):HS8-HS-12.
ABSTRACT: Susac syndrome, a relatively rare autoimmune endotheliopathy affecting the brain, retina, and inner ear, is three times more common in women than in men. The three main characteristics are encephalopathy, partial or complete branch retinal artery occlusion, and low-frequency hearing loss. Diagnosis is made via patient history, clinical presentation, and specialized tests including MRI, fluorescein angiography, and audiogram. Although Susac syndrome is self-limiting and sometimes resolves spontaneously, early and aggressive treatment is recommended to prevent or minimize potential irreversible damage to the brain, retina, and inner ear. Usual treatment involves immunosuppression with high-dose glucocorticoids and IV immunoglobulin. Other therapies include mycophenolate mofetil, rituximab, cyclophosphamide, azathioprine, and anti–tumor necrosis factor agents. Studies are examining other treatment options, such as monoclonal antibodies.
In 1979, John Susac, MD, and two colleagues reported the cases of two patients who presented with encephalopathy, hearing loss, and retinal microangiopathy. This condition was later named Susac syndrome (SuS).1 Today, despite continued research, little is known about its cause. SuS is relatively rare, debilitating, and complex.
SuS, an autoimmune endotheliopathy, injures vessels in the brain, retina, and inner ear. In its classic presentation, it is characterized by a clinical triad of 1) encephalopathy; 2) branch retinal artery occlusion (BRAO; partial or complete blockage of the arteries and capillaries supplying blood to the retina); and 3) low-frequency sensorineural hearing loss.2 SuS usually occurs in persons aged 20 to 40 years, but it has been reported in those ranging in age from 9 to 72 years. Women are three times more likely than men to develop SuS. Although a specific ethnicity has not been identified, white populations may be at higher risk. No genetic or hereditary links have been identified.3
Clinical Presentation
The encephalopathy, BRAO, and hearing loss associated with SuS typically are not all present at disease onset, and some patients do not develop all three. The multisystem involvement of SuS commonly imitates other disorders, significantly delaying the time to diagnosis.2 In SuS, encephalopathy manifests as headache, motor deficiencies, sensor deficiencies, aphasia, cognitive impairment, and urinary insufficiency. The hearing loss is usually bilateral. The BRAO may be extensive or subtle, and unilateral or bilateral.4
In more than 50% of SuS patients, the initial symptom is headache (sometimes involving features of migraine). Additional symptoms of brain involvement may occur, such as slurred speech, decreased executive function or memory loss, and gait disturbance.5 The development of behavioral and psychiatric symptoms, such as paranoia or personality disorder, has also been observed. Symptom presentation varies among patients.
The retinopathy of SuS is characterized by multiple peripheral BRAOs, which will be seen on ophthalmoscopic examination or retinal fluorescein angiography (FA). The restricted blood flow causes retinal injury and may lead to permanent vision loss. Some patients may not notice visual changes; others may experience scotoma, blurred vision, or partial or complete vision loss.6
Hearing loss related to SuS is often acute, unilateral or bilateral, asymmetrical, and associated with tinnitus, vertigo, nausea, vomiting, nystagmus, and unsteady gait. It is thought to be caused by restricted blood flow through the small blood vessels supplying the cochlea, which ultimately leads to infarction of the cochlear apex.6 The extent of hearing loss ranges from mild to severe, and in some cases it is detectable only by audiogram. Vertigo occurs when the vestibular apparatus is affected. Some patients recover significantly after the acute phase of SuS, but in severe cases deafness may result and cochlear implants may be needed.7
There is great variability among patients in the natural disease course, including the timing, extent, and duration of initial peak disease severity. SuS is self-limiting and sometimes resolves spontaneously. Some patients have a mild, relatively brief disease course (sometimes lasting <1 year), with fully reversible ischemic dysfunction in the brain, retina, and/or inner ear, and sustain little or no noticeable residual damage.8 Other patients have severe, prolonged disease lasting several years and will sustain devastating permanent damage from microinfarctions in all organs involved.8
Diagnosis
SuS is diagnosed based primarily on the patient’s clinical presentation and history, documentation of BRAO, presence of typical features on imaging (FA), audiogram results, and characteristic findings on cerebral MRI that help distinguish SuS from other inflammatory diseases, such as multiple sclerosis and acute disseminated encephalomyelitis.9
Brain involvement in a suspected SuS patient is determined by assessing symptoms such as alteration of consciousness, new cognitive or behavioral changes, new focal neurologic symptoms, and new headache. Additionally, at least one of the classic clinical findings located in the corpus callosum, such as “snowball” lesions, is present on MRI imaging.2,10
Retinal involvement is determined based on close ophthalmologic evaluation. The presence of at least one of the following is indicative of SuS: 1) at least one acute BRAO, 2) arterial-wall hyperfluoresence seen on FA, or 3) sectorial damage of the inner retinal layers from the retinal nerve fiber layer through the outer plexiform layer on optical coherence tomography. Additionally, the presence of visual-field deficits or scotoma may support the diagnosis of SuS.10-12
Vestibulocochlear involvement is assessed based on the presence of at least one of the following clinical manifestations: new tinnitus, hearing loss, or peripheral vertigo. These findings can be further confirmed by performing pure-tone or speech audiogram testing, preferably for low- or mid-tone frequencies.2 Definitive diagnosis of SuS may be made in patients fulfilling all of the above criteria; however, if only one or two criteria are met, a patient may be classified as having probable SuS and may require further investigation.
Treatment
Symptom presentation is unique in each patient, and the severity of symptoms may vary among the three organs involved. Therefore, the approach to treatment depends on which component of the clinical triad poses the greatest threat. In general, early and aggressive treatment is recommended to prevent or minimize potential irreversible damage to the brain, retina, and inner ear.
In most patients, treatment begins with immunosuppressive therapy using IV glucocorticoids (mainly methylprednisolone) followed by oral corticosteroids (prednisone) and IV immunoglobulin (IVIG) pulsed every 4 weeks for several months.5 The frequency and duration of treatment may vary based on the severity of the encephalopathy. The implementation of glucocorticoids is highly beneficial, and they should be considered as first-line therapy. A high dose of IV methylprednisolone administered during the acute phase of SuS has been shown to cause remission in the majority of patients. However, in as many as 10% of patients, such treatment is ineffective and may lead to further symptom progression.13,14 Therefore, it is recommended that all patients with encephalopathy are started on IVIG and/or mycophenolate mofetil (MMF) in addition to corticosteroids in order to prevent or minimize relapse during prednisone tapering and permit more rapid tapering of prednisone. For patients who experience breakthrough disease after adding MMF and rituximab, high-dose cyclophosphamide may be effective.11,12 However, if cyclophosphamide is contraindicated, azathioprine and IVIG therapy may be used, although a lesser effect has been demonstrated.15 Additionally, patients with extremely severe SuS and those who do not improve with standard therapy may benefit from plasma-exchange therapy.15 Studied dosages of the medications noted above are listed in TABLE 1.
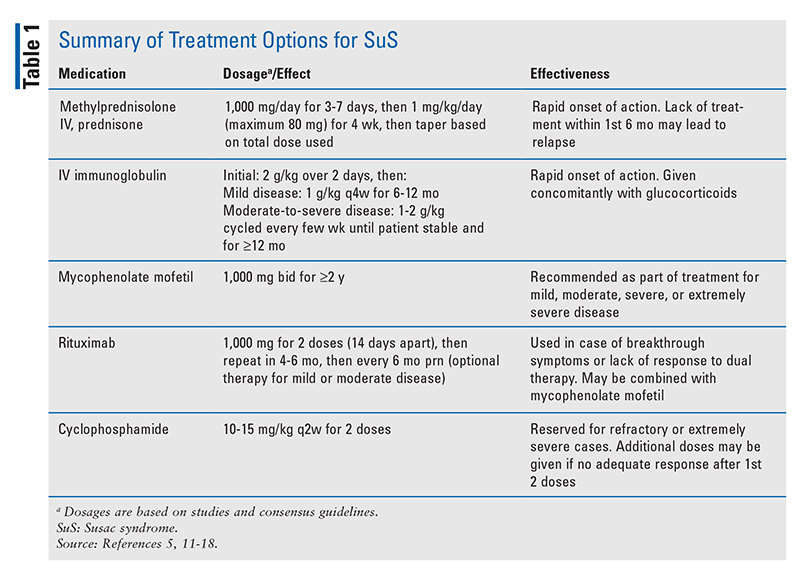
Duration of therapy depends strongly on the severity of presenting symptoms and the response to treatment. Methylprednisolone pulses are given for 3 to 7 days followed by prednisone for 4 weeks, after which therapy may be tapered slowly depending on the initial dosage and the duration.5 IVIG tapering begins when the patient is stable and improving on therapy, although a total course of up to 1 year may be needed, along with a 6-month taper (TABLE 1). MMF treatment usually continues for up to 2 years, whereas rituximab may be given every 6 months as needed.5
Other medications that have been investigated for the subacute disease phase are azathioprine, aspirin, and dipyridamole. Antithrombotic and anticoagulation agents may be considered as part of the SuS regimen in cases of visible retinal-vessel occlusion.9 Specific symptoms of SuS should resolve after initiation of high-dose steroids; however, they can be managed as needed. In SuS patients, migraines respond to triptans and topiramate, and severe hearing loss may be treated with cochlear implants, if desired.15
After initial diagnosis, the patient should be reassessed at 1 month and again at 3 months with visual-field testing, MRI, and FA.16 These tests also should be performed if the patient complains of new symptoms, and audiogram testing may be repeated if the patient complains of new hearing loss. Medical management is inadequate if MRI detects any disease activity (denoted by active enhancing lesions).16 Therapy is also inadequate if a new visual-field defect or arterial-wall damage is seen on FA, and maintenance therapy should be continued. When the MRI is inactive, FA is inactive, and visual fields are stable, the patient may be considered to be in remission, and one of the drugs may be withdrawn.17,18 With continued remission, other medications may be tapered slowly until the patient is weaned off all medications.
Several agents have been suggested as alternative treatments for SuS, such as monoclonal antibodies and anti–tumor necrosis factor (anti-TNF) agents, based on their applicability to other diseases that are similar to SuS.19 Currently, however, no studies on these agents’ use in SuS have been published; therefore, they are not recommended. Although no clinical trials on the treatment of SuS are ongoing, there is a clear need for more streamlined treatment options and better management of affected patients.19
Disease Prognosis
SuS presents with varying levels of severity and differs greatly in its response to treatment in individual patients. Therefore, it is difficult to predict long-term outcomes and quality of life. Whereas some individuals will experience symptoms only once in their lifetime, others may have several episodes before the symptoms resolve, and still others may have a chronic relapsing course with frequent flare-ups and remissions.13 The usual disease course is 2 to 4 years, but the course may be shorter or longer.
Although certain patients recover from SuS with minimal to no long-term complications, others continue to have cognitive deficits, gait disturbance, or hearing loss (vision is usually not seriously impaired).20 Most patients recover from SuS, but they may experience some disability that may impair quality of life. There are only a few reported cases of patients who have died from complications of SuS.21
Conclusion
SuS is a relatively rare, debilitating, and complex disease. Its severity ranges from mild to severe, and its presenting symptoms differ widely among patients. Although no definitive treatment has been established, there is sufficient evidence to select an appropriate course of action based on the patient’s presenting symptoms. Immunosuppressive therapy—mainly with corticosteroids such as prednisone and methylprednisolone—and IVIG have the most efficacy in suppressing SuS and preventing relapses. Other immunosuppressive agents, such as MMF and cyclophosphamide, may be used in varying combinations in patients who are unresponsive, have severe or extremely severe disease, or relapse on dual therapy. Although much current research centers on treatment with monoclonal antibodies and anti-TNF agents, the use of rituximab as an adjunct to first-line treatment is preferred based on available studies. Some patients have complete disease remission, whereas others experience periods of relapse. Pharmacist involvement is crucial for patients with SuS, not only for helping identify the disease and its presenting symptoms but also for managing therapy and providing current data on emerging treatments.
REFERENCES
1. Susac JO. Susac’s syndrome: the triad of microangiopathy of the brain and retina with hearing loss in women. Neurology. 1994;44(4):591-593.
2. Rennebohm R, Susac JO, Egan RA, Daroff RB. Susac’s syndrome—update. J Neurol Sci. 2010;299(1-2):86-91.
3. Seifert-Held T, Langner-Wegscheider BJ, Komposch M, et al. Susac’s syndrome: clinical course and epidemiology in a Central European population. Int J Neurosci. 2017;127(9):776-780.
4. Susac JO, Murtagh FR, Egan RA, et al. MRI findings in Susac’s syndrome. Neurology. 2003;61(12):1783-1787.
5. Rennebohm RM, Asdaghi N, Srivastava S, Gertner E. Guidelines for treatment of Susac syndrome—an update. Int J Stroke. 2020;15(5):484-494.
6. Egan RA, Nguyen TH, Gass JDM, et al. Retinal arterial wall plaques in Susac syndrome. Am J Ophthalmol. 2003;135(4):483-486.
7. Susac JO. Susac’s syndrome: the triad of microangiopathy of the brain and retina with hearing loss in young women. Neurology. 1994;44(4):591-593.
8. Dörr J, Krautwald S, Wildemann B, et al. Characteristics of Susac syndrome: a review of all reported cases. Nat Rev Neurol. 2013;9(6):307-316.
9. Kleffner I, Dörr J, Ringelstein M, et al. Diagnostic criteria for Susac syndrome. J Neurol Neurosurg Psychiatry. 2016;87(12):1287-1295.
10. Egan RA, Hills WL, Susac JO. Gass plaques and fluorescein leakage in Susac syndrome. J Neurol Sci. 2010;299(1-2):97-100.
11. Ringelstein M, Albrecht P, Kleffner I, et al. Retinal pathology in Susac syndrome detected by spectral-domain optical coherence tomography. Neurology. 2015;85(7):610-618.
12. Brandt AU, Oberwahrenbrock T, Costello F, et al. Retinal lesion evolution in Susac syndrome. Retina. 2016;36(2):366-374.
13. Rennebohm RM, Susac JO. Treatment of Susac’s syndrome. J Neurol Sci. 2007;257(1-2):215-220.
14. Rennebohm RM, Egan RA, Susac JO. Treatment of Susac’s syndrome. Curr Treat Options Neurol. 2008;10(1):67-74.
15. Mateen FJ, Zubkov AY, Muralidharan R, et al. Susac syndrome: clinical characteristics and treatment in 29 new cases. Eur J Neurol. 2012;19(6):800-811.
16. Vodopivec I, Venna N, Rizzo JF III, Prasad S. Clinical features, diagnostic findings, and treatment of Susac syndrome: a case series. J Neurol Sci. 2015;357(1-2):50-57.
17. Vodopivec I, Prasad S. Treatment of Susac syndrome. Curr Treat Options Neurol. 2016;18(1):3-10.
18. Egan RA. Diagnostic criteria and treatment algorithm for Susac syndrome. J Neuroophthalmol. 2019;39(1):60-67.
19. Hardy TA, Garsia RJ, Halmagyi GM, et al. Tumour necrosis factor (TNF) inhibitor therapy in Susac’s syndrome. J Neurol Sci. 2011;302(1-2):126-128.
20. Aubart-Cohen F, Klein I, Alexandra JF, et al. Long-term outcome in Susac syndrome. Medicine (Baltimore). 2007;86(2):93-102.
21. Saux A, Niango G, Charif M, et al. Susac’s syndrome, a rare, potentially severe or lethal neurological disease. J Neurol Sci. 2010;297(1-2):71-73.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
To comment on this article, contact rdavidson@uspharmacist.com.



