US Pharm. 2022;47(2):4.
ABSTRACT: Homozygous familial hypercholesterolemia (HoFH) is an inherited disorder characterized by dangerously elevated low-density lipoprotein (LDL) levels. Untreated, it leads to the rapid development of atherosclerosis. Treatment consists of both pharmacologic and nonpharmacologic therapies. It is difficult to reach therapeutic goals in this patient population due to the need for drastic reductions in LDL cholesterol levels. Fortunately, newer therapies make reaching these goals more feasible. Pharmacists can play a large part in educating patients with HoFH about the importance of treatment, as well as ensuring proper drug dosing, administration, and monitoring.
Homozygous familial hypercholesterolemia (HoFH) is an autosomal-dominant disorder involving disturbances in lipid metabolism that lead to high levels of low-density lipoprotein (LDL).1 Prolonged elevations in LDL cholesterol (LDL-C) levels increase a person’s risk of developing atherosclerotic cardiovascular disease (ASCVD) and experiencing premature and recurrent coronary events.2,3 Because patients with HoFH have inherited two LDL-C–raising gene mutations, they experience a more severe form of this disease, with LDL-C levels of 400 mg/dL or higher.4,5 This disorder presents with severe hypercholesterolemia from birth and onset of cardiovascular disease during early childhood.6 Some of the physical manifestations of the disorder include the development of xanthoma and xanthelasma, cholesterol deposits under the skin and on or around the eyelids.6
Studies have shown that HoFH is more common than previously thought.7 Although there are no data on the prevalence of HoFH in the United States, one study in the Netherlands found that the prevalence was approximately 1 in 300,000.7 Because the U.S. is a racially and ethnically diverse country, this rate, which has been extrapolated from a primarily Caucasian population, may not accurately predict the prevalence rate. HoFH is highest among whites, blacks, and other Hispanics, and lowest among Mexican Americans and other races.8 HoFH is a treatable condition that must be diagnosed early on in life to prevent rapid atherosclerotic development and premature coronary events. Unfortunately, it is consistently underdiagnosed and undertreated, with patients often not diagnosed until after their first coronary event.9
Pathophysiology
In approximately 90% of cases, HoFH is caused by mutations in the LDL receptor (LDLR) gene that encodes the LDL receptor.10 The LDLR gene is responsible for the clearance of a majority of LDL from the plasma.11 An LDLR mutation can affect the synthesis, transport, binding affinity, internalization, or recycling of LDL receptors.11 Mutations in other genes, specifically apolipoprotein B (APOB), proprotein convertase subtilisin kexin type 9 (PCSK9), and LDL receptor adaptor protein 1 (LDLRAP1), have also been identified as causing HoFH.10,11 The APOB gene mutation comprises about 5% of all cases of HoFH.10 Mutations in the APOB gene lead to structural changes in the apoB ligand, which interferes with the binding of the LDL particle to the LDL receptor, leading to increased circulating LDL-C.10,11 Only about 1% of all HoFH cases are linked to mutations in the PCSK9 gene, which prevents the LDLR from being recycled and promotes its degradation.10 With fewer LDL receptors available to bind LDL-C, circulating LDL-C levels increase.10 LDLRAP1 mutations lead to the formation of a nonfunctional LDLRAP1.11 Without this functioning protein, LDL can still bind to the LDLR, but it cannot be transported into the cell.11 This mutation leads to a rare autosomal recessive disorder.11
Diagnosis
It is estimated that more than 90% of individuals with HoFH remain undiagnosed due to a lack of standardized diagnostic criteria.12,13 The American Heart Association has proposed a simple clinical classification for HoFH with the goal of better recognition of the disorder.11,12 TABLE 1 provides this classification. Although genetic testing has the potential to improve diagnoses, data received from the U.S. CASCADE FH registry indicate that genetic testing was used in only 3.9% of individuals who were diagnosed with FH.13 Cascade screening, which involves testing all first-degree relatives of patients who have been diagnosed with HoFH for elevated LDL-C or a known genetic mutation, can help increase early recognition and treatment of HoFH.14

Treatment
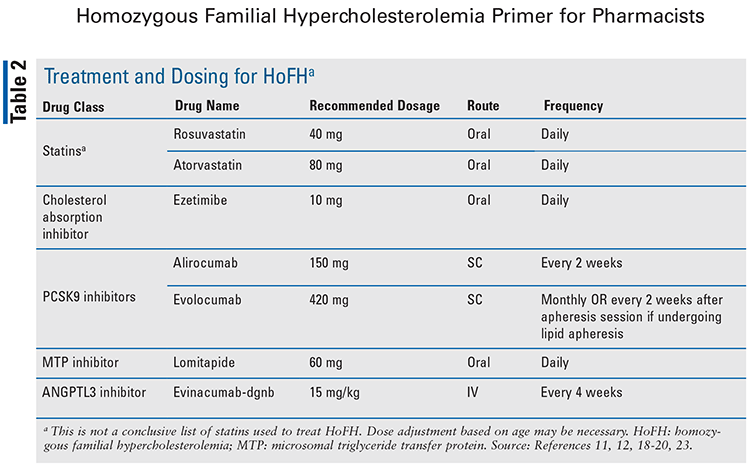
Every patient who is diagnosed with HoFH should be referred to a lipid specialist for advanced management of the condition.5 The primary goal of treatment for HoFH is to reduce mortality and prevent ASCVD progression.5 The American Heart Association/American College of Cardiology recommends initiation of a high-intensity statin to lower LDL-C levels by a minimum of 50% in high-risk and very high–risk patients.4 The European Atherosclerosis Society guidelines state that goal LDL-C levels in patients with HoFH are <100 mg/dL for adults without clinical ASCVD, <70 mg/dL for adults with clinical ASCVD, and <130 mg/dL in children.16 Although HoFH cannot be adequately controlled with lifestyle modifications alone, they should be recommended adjunct to pharmacologic therapy, as these patients have a very high baseline risk.15,16 Pharmacologic therapies that are currently being used to treat HoFH include statins, a cholesterol absorption inhibitor, PCSK9 inhibitors, a microsomal triglyceride transfer protein (MTP) inhibitor, and an angiopoietin-like 3 (ANGPTL3) inhibitor.4,5 TABLE 2 provides a list of these drug treatments with dosages for use in HoFH.
Lipoprotein apheresis and liver transplantation are two nonpharmacologic therapies that are used in patients who are unresponsive to pharmacologic and dietary management.4,16 Lipoprotein apheresis is an extracorporeal treatment than removes APOB-containing lipoproteins from the circulation, leading to a rapid reduction in LDL-C levels.5,11 Even though a single treatment can reduce LDL-C levels by 50% to 60%, there are many disadvantages associated with this treatment, including problems with venous access, transient hypotension, fatigue, bleeding, hypocalcemia, and iron deficiency.5 Patient inconvenience is also a problem, as this is a weekly or biweekly treatment that takes a few hours to complete.4,5 Liver transplantation is the only curative option for patients with HoFH, correcting the molecular defect in the liver that causes LDL-C elevations.16 There are many disadvantages associated with liver transplantation, including difficulty in finding a donor, organ rejection, and lifelong immunosuppressive therapy.16
Lifestyle Modifications
Dietary changes should include a reduction in the consumption of trans fats, saturated fats, and foods with high cholesterol content.9 Decreasing sedentary behaviors, such as sitting or lying down, while increasing physical activity has been found to decrease ASCVD risk.3 Other important lifestyle modifications found to decrease ASCVD risk include tobacco cessation, alcohol moderation, and weight loss in those considered overweight or obese.3 Achieving treatment goals in patients with type 2 diabetes mellitus or high blood pressure can reduce ASCVD risk.3
Statins
Statins are a class of medications that inhibit the enzyme HMG-CoA reductase, which is the rate-limiting step in cholesterol biosynthesis.4 This leads to a reduction in hepatic LDL-C, triggering a compensatory upregulation in LDL receptors that reuptake LDL-C from the blood, reducing plasma LDL-C levels.4 Statins should be initiated at diagnosis or as early as possible; however, individual statin drugs have different FDA-approved ages ranging from 7 to 10 years and older.11,17 Even so, the American Academy of Pediatrics recommends starting pharmacologic treatment in patients younger than age 8 years if they persistently have LDL-C levels >500 mg/dL.17 Statins lead to reductions of 10% to 25% in LDL-C levels in the majority of patients with HoFH and even reduce LDL-C levels in patients who have LDLR-negative HoFH.11,15 Despite only leading to modest reductions in LDL-C levels, statins continue to be the mainstay of treatment, as they have been found to reduce cardiovascular and all-cause mortality.16 Adverse events reported with statin use include headache, dyspepsia, and myalgias.11
Cholesterol Absorption Inhibitor
Ezetimibe is an oral, once-daily medication that works by inhibiting cholesterol transport in the intestines by blocking the cholesterol transport protein Niemann-Pick C1-like protein.4,11 Blocking this protein leads to a reduction in LDL-C levels in the liver, triggering an upregulation of LDL receptors and a subsequent reuptake of LDL-C from the blood.4,11 It is FDA approved for use in patients with HoFH in combination with a high-intensity statin to reduce total cholesterol and LDL-C levels.5 When added to statin therapy, it was found to reduce LDL-C levels by an additional 10% to 15%.11 It is generally well tolerated, with reports of nasopharyngitis, myalgia, upper respiratory tract infection, arthralgia, and diarrhea when used in combination with a statin.5 Due to its lack of dependence on LDLR expression for its effect, ezetimibe may be advantageous in patients with HoFH who are LDLR-negative.14
PCSK9 Inhibitors
PCSK9 inhibitors are a class of monoclonal antibodies administered subcutaneously that bind to PCSK9, an enzyme that regulates the levels of LDL receptors and prevents it from interacting with the LDL receptor.11,12 By preventing this interaction, PCSK9 inhibitors increase the recycling of LDL receptors to the cell surface of hepatocytes and increases the clearance of LDL-C from the plasma.11,12 Currently, there are two approved medications, alirocumab and evolocumab. In the ODYSSEY HoFH study, 69 patients were randomized 2:1 to receive alirocumab 150 mg or placebo every 2 weeks. LDL-C was reduced from baseline by 26.9% in the alirocumab group compared with an increase of 8.6% from baseline in the placebo group.18 LDL-C reduction was highly dependent on residual LDL receptor activity, with treatment being ineffective in one-third of patients homozygous for LDL receptor, PCSK9, or LDLRAP1.4,18 Adverse effects were reported in 44.4% of patients receiving alirocumab, with upper respiratory tract infection, headache, and diarrhea being the most common.18
In the TAUSSIG trial, long-term safety and efficacy data for evolocumab in patients with HoFH and HeFH were assessed.19 Of the 300 participants, 106 were patients with HoFH.19 Participants received either evolocumab 420 mg once-monthly if they were not undergoing lipid apheresis or evolocumab 420 mg every 2 weeks if they were undergoing lipid apheresis.19 Mean percentage changes in LDL-C were –21.2% at Week 12 and –24.0% at Week 216 for patients with HoFH.19 Adverse effects were reported in 89.3% of participants receiving evolocumab, with nasopharyngitis, influenza, upper respiratory tract infection, headache, myalgia, and diarrhea being the most common.19
MTP Inhibitor
Lomitapide is an oral inhibitor of MTP.16 MTP is responsible for transferring triglycerides and phospholipids onto chylomicron and very-low-density lipoproteins (VLDL) during their assembly in the intestines and the liver, respectively.16 By inhibiting MTP, lomitapide causes a reduction in the secretion of chylomicrons and VLDL, which leads to a reduction in LDL-C levels.16 It is FDA approved for use as an adjunct to a low-fat diet and other lipid-lowering therapies to decrease LDL-C and APOB in patients with HoFH.5 Lomitapide can be used in patients who have a lack of LDLR expression, because its mechanism of action is independent of LDLR function.12 In a single-arm, open-label, phase III trial of lomitapide in patients with HoFH, lomitapide reduced LDL-C levels by 50% from baseline at Week 26.20 At Week 126, the mean percent change in LDL-C was –45.5%, in 19 patients who were enrolled in an extension trial, with this reduction being maintained through the end of the 282-week trial.21
A survival-benefit analysis was conducted that found starting lomitapide at age 18 years could increase life expectancy by 11.2 years and delay the time to first major adverse cardiovascular event by 5.7 years in patients with HoFH.22 Lomitapide is a CYP3A4 substrate, and concomitant use of strong and moderate CYP3A4 inhibitors is contraindicated.5 The most common adverse events were gastrointestinal symptoms, including nausea, flatulence, and diarrhea, and liver fat accumulation.16,20
During the extension trial, elevations in alanine aminotransferase or aspartate aminotransferase greater than five times the upper limit of normal were reported in four patients (21.1%).21 However, these were typically associated with concomitant use of CYP450 3A4 inhibitors or excess alcohol use, and these events were managed by discontinuing the offending agent, reducing or suspending the use of lomitapide, and reintroducing lomitapide after normalization of transaminases.21 A Risk Evaluation and Mitigation Strategy (REMS) program and registry has been established for this drug to monitor for hepatotoxicity, cirrhosis, and insulin resistance.11,12 Prescribers and pharmacies must be specially certified in the REMS program to prescribe and dispense this medication.11,12
ANGPTL3 Inhibitor
Evinacumab-dgnb, the most recently approved treatment for HoFH, is a fully human monoclonal antibody administered IV every 4 weeks.4,23 It binds to and inhibits ANGPTL3, an endogenous inhibitor of lipoprotein and endothelial lipase, which is known to increase the levels of triglycerides and other lipids.23 Inhibition of ANGPTL3 leads to a reduction in VLDL lipid content and particle size, which improves their clearance from the circulation.4 The reduction in VLDL remnants leads to a reduction in LDL production and circulating LDL-C levels.4 In the Evinacumab Lipid Studies in Patients with Homozygous Familial Hypercholesterolemia trial, patients in the evinacumab group had a 47.1% LDL-C reduction from baseline at Week 24 compared with a 1.9% increase in the placebo group.23
In patients who had <15% LDLR activity, evinacumab lowered LDL-C levels by 43.4% compared with placebo, which increased LDL-C levels by 16.2%.23 Additionally, evinacumab reduced apoB levels by 41.4% from baseline at Week 24.23 HDL-C levels were reduced by 29.6% from baseline at Week 24 in the evinacumab group compared with an increase of 0.8% in the placebo group.23 A higher percentage of patients reported adverse events in the placebo group than the evinacumab group (81% vs. 66%, respectively).23 The most common adverse events reported were nasopharyngitis, influenza-like illness, headache, and rhinorrhea.23
Conclusion
HoFH is an important disorder for pharmacists to be aware of since it is associated with rapid development of ASCVD and premature coronary events, and yet it remains underdiagnosed and undertreated. Pharmacists can play a role in addressing the need for strict adherence to cholesterol-lowering therapies to achieve maximum efficacy from treatment. Additionally, pharmacists can monitor for drug interactions and adverse effects, which could contribute to poor outcomes. Newer therapies continue to emerge that make reaching LDL-C goals in patients with HoFH possible, and pharmacists can provide the necessary patient education to help further improve outcomes.
REFERENCES
1. Vallejo-Vaz AJ, Seshasai SRK, Cole D, et al. Familial hypercholesterolaemia: a global call to arms. Atherosclerosis. 2015;243(1):257-259.
2. Perak AM, Ning H, de Ferranti SD, et al. Long-term risk of atherosclerotic cardiovascular disease in US adults with the familial hypercholesterolemia phenotype. Circulation. 2016;134(1):9-19.
3. Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. Circulation. 2019;140(11):e596-e646.
4. Rosenson RS. Existing and emerging therapies for the treatment of familial hypercholesterolemia. J Lipid Res. 2021;62:100060.
5. Lloyd-Jones DM, Morris PB, Ballantyne CM, et al. 2017 focused update of the 2016 ACC Expert Consensus Decision Pathway on the role of non-statin therapies for LDL-cholesterol lowering in the management of atherosclerotic cardiovascular disease risk: a report of the American College of Cardiology Task Force on Expert Consensus Decision Pathways. J Am Coll Cardiol. 2017;70(14):1785-1822.
6. Kolansky DM, Cuchel M, Clark BJ, et al. Longitudinal evaluation and assessment of cardiovascular disease in patients with homozygous familial hypercholesterolemia. Am J Cardiol. 2008;102(11):1438-1443.
7. Sjouke B, Kusters DM, Kindt I, et al. Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: prevalencxe, genotype–phenotype relationship, and clinical outcome. Eur Heart J. 2015;36(9):560-565.
8. De Ferranti SD, Rodday AM, Mendelson MM, et al. Prevalence of familial hypercholesterolemia in the 1999 to 2012 United States National Health and Nutrition Examination Surveys (NHANES). Circulation. 2016;133(11):1067-1072.
9. Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34(45):3478-3490a.
10. Chemello K, Garcia-Nafria J, Gallo A, et al. Lipoprotein metabolism in familial hypercholesterolemia. J Lipid Res. 2021;100062.
11. Gidding SS, Champagne MA, de Ferranti SD, et al. The agenda of familial hypercholesterolemia. Circulation. 2015;132(22):2167-2192.
12. Lui DTW, Lee ACH, Tan KCB. Management of familial hypercholesterolemia: current status and future perspectives. J Endocr Soc. 2021;5(1):bvaa122.
13. Sturm AC, Knowles JW, Gidding SS, et al. Clinical genetic testing for familial hypercholesterolemia: JACC Scientific Expert Panel. J Am Coll Cardiol. 2018;72(6):662-680.
14. Raal FJ, Santos RD. Homozygous familial hypercholesterolemia: current perspectives on diagnosis and treatment. Atherosclerosis. 2012;223(2):262-268.
15. Turgeon RD, Barry AR, Pearson GJ. Familial hypercholesterolemia: review of diagnosis, screening, and treatment. Can Fam Physician. 2016;62(1):32-37.
16. Cuchel M, Bruckert E, Ginsberg HN, et al. Homozygous familial hypercholesterolemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolemia of the European Atherosclerosis Society. Eur Heart J. 2014;35(32):2146-2157.
17. Eiland LS, Luttrell PK. Use of statins for dyslipidemia in the pediatric population. J Pediatr Pharmacol Ther. 2010;15(3):160-172.
18. Blom DJ, Harada-Shiba M, Rubba P, et al. Efficacy and safety of alirocumab in adults with homozygous familial hypercholesterolemia: the ODYSSEY HoFH trial. J Am Coll Cardiol. 2020;76(2):131-142.
19. Santos RD, Stein EA, Hovingh GK, et al. Long-term evolocumab in patients with familial hypercholesterolemia. J Am Coll Cardiol. 2020;75(6):565-574.
20. Cuchel M, Meagher EA, du Toit Theron H, et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013;381(9860):40-46.
21. Blom DJ, Averna MR, Meagher EA, et al. Long-term efficacy and safety of the microsomal triglyceride transfer protein inhibitor lomitapide in patients with homozygous familial hypercholesterolemia. Circulation. 2017;136(3):332-335.
22. Leipold R, Raal F, Ishak J, et al. The effect of lomitapide on cardiovascular outcome measures in homozygous familial hypercholesterolemia: a modelling analysis. Eur J Prev Cardiol. 2017;24(17):1843-1850.
23. Raal FJ, Rosenson RS, Reeskamp LF, et al. Evinacumab for homozygous familial hypercholesterolemia. N Engl J Med. 2020;383:711-720.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
To comment on this article, contact rdavidson@uspharmacist.com.


