US Pharm. 2024;49(6):HS11-HS16.
ABSTRACT: Despite relatively low mortality, prostate cancer (PCa) survivability decreases substantially with metastasis. Conventional treatment modalities such as androgen deprivation therapy and antineoplastic therapy do not always achieve optimal outcomes. Risk factors for developing PCa include insulin resistance and metabolic syndrome. Incretin-based pharmacotherapy, a well-established approach for managing diabetes and obesity, may be beneficial in PCa and mitigate current therapeutic challenges. Treatment with glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-IV inhibitors may reduce PCa risk, inhibit disease progression, or circumvent therapeutic resistance by improving overall metabolic health, increasing sensitivity to antiandrogen agents, and amplifying the efficacy of antineoplastic drugs.
Over the past decade, the use of incretin-based pharmacotherapies such as glucagon-like peptide-1 receptor agonists (GLP1RA) and dipeptidyl peptidase-IV inhibitors (DPP-IVi) to promote weight loss and improve glycemic control has skyrocketed.1,2 Currently, three GLP1RA are approved by the FDA as antiobesity agents: liraglutide (Saxenda), semaglutide (Wegovy), and tirzepatide (Zepbound). These and other GLP1RA medications signal brain regions that regulate feeding, such as the hypothalamus and hindbrain, to reduce food intake, appetite, and hunger.3 In the gastrointestinal (GI) tract, they promote fullness and satiety by delaying gastric emptying and reducing intestinal motility.3 As incretin mimetics, they also promote glucose homeostasis by increasing pancreatic glucose-stimulated insulin secretion (GSIS) and decreasing glucagon secretion.3 Unlike GLP1RA, DPP-IVi are weight-neutral (i.e., do not promote weight loss).4 DPP-IVi prevent the breakdown of both GLP-1 and glucose-dependent insulinotropic polypeptide by the ubiquitous enzyme DPP-IV.4 As a result, circulating GLP-1 and gastric inhibitory polypeptide levels rise to promote pancreatic insulin secretion and reduce postprandial and fasting hyperglycemia. FDA-approved drugs in this class include sitagliptin, vildagliptin, saxagliptin, alogliptin, and linagliptin.
GLP1RA and DPP-IVi may also be effective for conditions associated with obesity and insulin resistance (IR), including prostate cancer (PCa).5 PCa involves proliferation of malignant glandular cells (i.e., adenocarcinoma) in the prostate gland, a male reproductive organ largely responsible for producing seminal fluid.6 PCa is the second most common cancer occurring in men and the fifth leading cause of death worldwide.7 According to the National Cancer Institute Surveillance, Epidemiology, and End Results Program, 288,300 new PCa cases were diagnosed in 2023, accounting for ~15% of all new cancer diagnoses.8 In 2020, ~3.5 million American men were living with PCa.8 Mortality is extremely low, as the estimated 5-year survival rate is 97.5%. However, survivability decreases significantly if PCa metastasizes, with a 5-year survival rate of only 34.1% and ~85% of metastatic cases involving bone.8,9
Multiple lines of evidence show antitumor effects of GLP1RA and DPP-IVi in PCa through actions that improve metabolic health and involve sensitization to antiandrogen agents and amplification of antineoplastic therapies.5,10 Some of the most compelling evidence linking incretin-based pharmacotherapy to reduced PCa risk comes from observations that the GLP1R is expressed in primary human PCa tissues in vitro, with the highest expression observed in malignancies that are androgen-sensitive (vs. androgen-independent).11 Moreover, treatment with GLP1RA reduces PCa cell proliferation in vitro and growth of transplanted PCa cell lines in vivo.12 This article will discuss the underlying mechanisms for a potential benefit of incretin-based pharmacotherapy in PCa.
PCa Pathophysiology
Androgen-Dependent Androgen Receptor (AR) Signaling: Androgens such as testosterone and its active metabolite dihydrotestosterone (DHT) are steroid hormones that regulate the development, differentiation, and survival of prostate gland cells by binding to the AR.13 The AR is a ligand-dependent nuclear transcription factor located in the cytoplasm when inactive.14 AR signaling is classified as genomic or nongenomic.14 With genomic AR signaling, androgen-bound (i.e., active) AR translocates to the nucleus, where it regulates the transcription of target genes.14 In contrast, nongenomic AR signaling does not require androgen-bound AR translocation to the nucleus and binding to DNA.14 Instead, AR signaling is regulated by nonsteroidal membrane-bound and cytosolic effectors, including the phosphoinositide 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway, that are activated by growth factors, chemokines, and cytokines.14,15 However, the AR can undergo posttranslational modification (e.g., phosphorylation, acetylation, methylation, ubiquitination) and eventually translocate to the nucleus to regulate gene transcription.14 AKT, a serine/threonine kinase, may serve as a junction between genomic and nongenomic AR signaling, which suggests that it compensates in response to selective pressures.14 In PCa, hyperactive AR signaling supports cell growth, proliferation, and survival partly by reprogramming mitochondrial metabolism as well as by activating tumorigenic intracellular signaling cascades.14,16 Complex and synergistic crosstalk between the PI3K/AKT/mTOR pathway and many other signal transduction pathways further promotes disease progression and contributes to therapeutic resistance.17
Androgen-Independent AR Signaling: PCa can develop into androgen-independent PCa or castration-resistant PCa, in which there is disease progression despite serum testosterone levels <20 ng/dL.18 Up to 20% of PCa cases progress to castration-resistant PCa within 5 years of diagnosis.19 As the tumor becomes more aggressive through adaptive responses, it relies less on AR signaling and more on alternative activated pathways.14 With androgen-independent signaling, genomic signaling is triggered via phosphorylation mediated by various cytoplasmic factors, including AKT, and nongenomic signaling is promoted by activation of the PI3K/AKT/mTOR pathway.14,15 Oncogenic adaptations include hyperactivation of this pathway and, in 20% of cases, loss of the tumor suppressor gene phosphatase and tensin homologue, which acts as a gatekeeper of the PI3/AKT/mTOR pathway via dephosphorylation.14,17 Additional deregulated signaling pathways contributing to cell growth, proliferation, differentiation, transformation, and survival, as well as inhibited apoptosis, include nuclear factor kappa B, Janus kinase/signal transducers and activators of transcription, mitogen-activated protein kinase (MAPK), transcription growth factor-beta/mothers against decapentaplegic, and Wnt.20 Furthermore, metastatic PCa is associated with the conversion of metabolic signaling pathways governed by the master cellular energy sensors mTOR and AMP-activated protein kinase (AMPK).21 Whereas circulating cancer cells display decreased mTOR and increased AMPK activities, these profiles are reversed after metastatic onset to facilitate mTOR-dependent tumor growth.21
Tumor Microenvironment: The PCa tumor microenvironment includes endothelial, epithelial, immune, stromal, neural crest, and cancer cells that release tumorigenic chemokines, cytokines, extracellular matrices, and matrix-degrading enzymes.22 In addition to interacting with each other, these cells interact with multiple soluble factors and proteins to create an immunosuppressive network that further fuels tumor progression and invasion.22 Hallmarks include hypoxia and associated stimulation of the angiogenic factors vascular endothelial growth factor and angiopoietin 1; increased glycolysis, lactate metabolism, acidification, and oxidative stress; and immunosuppression that helps PCa cells evade cellular immune destruction.22
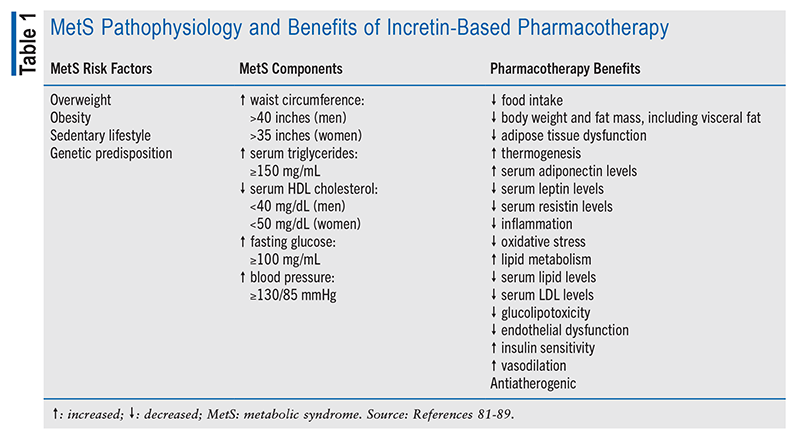
Metabolic Syndrome: Nonmodifiable risk factors for PCa include family history, ethnicity, and age. One major modifiable risk factor that is growing in prevalence is metabolic syndrome (MetS). MetS is a constellation of comorbidities characterized by IR, increased visceral fat, increased release of free fatty acids (that impair hepatic insulin clearance), and alterations in peripheral metabolism. Five criteria define MetS (TABLE 1), with the presence of three components required for a diagnosis. Abnormalities include abdominal adiposity, hypertension, IR, hyperinsulinemia, glucose intolerance, and dyslipidemia. MetS heightens the risk of developing a multitude of health problems, including atherosclerotic cardiovascular (CV) disease and type 2 diabetes (T2D). MetS, including the presence of visceral adiposity, contributes to increased serum levels of proinflammatory adipokines, such as interleukin-6, tumor necrosis factor-alpha, C-reactive protein, resistin, leptin, and macrophage chemoattractant protein-1.23,24 Reduced levels of adiponectin, interleukin-10, and insulin-like growth factor (IGF)-1/IGF binding proteins and increased oxidative stress further contribute to systemic inflammation and IR.25,26 Mounting evidence indicates that MetS, including IR, is associated with increased PCa risk.27 Elevated C-peptide, abdominal (or central) obesity (i.e., increased waist circumference), abnormal adipokine profile, and increased inflammation (or metaflammation) are associated with higher Gleason scores, which signify a worse prognosis.28,29 Multivariate analysis indicates that men with MetS have a twofold relative risk of developing PCa, which is further increased by being overweight or obese.30 Paradoxically, T2D is inversely related to PCa, with diabetic patients displaying reduced PCa risk.31
Limitations of Conventional Androgen Deprivation Therapy
Androgen deprivation therapy (ADT), a form of medical castration, is the hallmark approach to slowing down and controlling tumor growth.32 Androgen deprivation can be achieved by inhibiting androgen biosynthesis or blocking AR activation. ADT is the primary therapeutic option for high-risk, androgen-sensitive PCa, and it is commonly coupled with radiotherapy.33 FDA-approved ADT agents that suppress hormone production include gonadotropin-releasing hormone agonists (e.g., goserelin, buserelin, triptorelin, leuprolide) and antagonists (e.g., degarelix).34 Both drug classes lower testosterone to castrate levels (<50 ng/dL) by inhibiting luteinizing hormone and follicle-stimulating hormone.35 AR antagonists (e.g., enzalutamide, bicalutamide, abiraterone) completely block androgen-AR signaling, although they are only marginally beneficial when utilized as standalone treatments.32
Despite its efficacy, ADT has an extensive and potentially serious side-effect profile, including CV disorders (i.e., coronary heart disease, myocardial infarction, sudden cardiac death, stroke), bone fractures, metabolic dysfunction (i.e., IR, T2D), and impaired cognitive function.35 Therefore, the gold standard PCa treatment is radical prostatectomy; however, this intervention is accompanied by surgical morbidities, such as erectile dysfunction and incontinence. Also, ADT and AR antagonists are rendered ineffective in castration-resistant PCa. These challenges highlight the need for alternative and/or adjuvant therapies.
Benefits of Incretin-Based Pharmacotherapy in PCa
The pleiotropic effects of GLP1RA and DPP-IVi have a favorable impact on an array of physiological processes, several of which occur independently of insulin action.36,37 Incretin mimetics reduce body weight, improve postprandial glucose metabolism, augment muscle perfusion, increase whole-body glucose uptake, suppress hepatic glucose production, stimulate anti-inflammatory signaling, reduce endoplasmic reticulum stress, and induce autophagy.36 Moreover, these agents improve cardiac and respiratory function, reduce blood pressure, improve dyslipidemia, and prevent neurodegeneration.36-38 Beneficial incretin-independent effects of DPP-IVi include antihypertensive, anti-inflammatory, antiapoptotic, and immunomodulatory actions at the heart, kidneys, and vasculature.37 Regarding the risk of PCa, an inverse relationship between GLP1RA and DPP-IVi use (vs. other antidiabetic medications) has been observed.5,39
GLP-1 elicits its effects acutely by increasing intracellular cyclic AMP levels and subsequently activating various protein kinases, such as protein kinase A (PKA) and downstream AMPK.36 Additional downstream pathways stimulated by chronic GLP-1 exposure have been identified in multiple tissues, including pancreas, muscle (skeletal, cardiac, and smooth), liver, vasculature, kidney, adipose, and brain.36 In the brain, mTOR and AMPK signaling mediate the anorectic effect of GLP1RA.36,40 The food intake–suppressive effect of the GLP-1 analogue exendin-4 (Ex4; exenatide [Byetta]) requires mTOR activation in the ventromedial hypothalamus.40 Pharmacologic inhibition of glycolysis (i.e., activation of AMPK) in this brain region attenuates Ex4-induced anorexia, indicating that glucose metabolism and inhibition of AMPK are required for this effect.40 Confoundingly, the satiety-promoting activities of central mTOR and AMPK mirror those observed in metastatic PCa.21
GLP-1 also acts as a growth factor by promoting pancreatic beta-cell proliferation, survival, and neogenesis through actions at the epidermal growth factor receptor and downstream activation of PKA and PI3K/AKT signaling.41 By promoting pancreatic insulin secretion, GLP-1 indirectly stimulates hepatic glucose clearance and suppresses hepatic glucose production through PI3K/AKT and AMPK activation via the gut-pancreas-liver axis as well as through actions in the brain.42,43 With liraglutide, improved insulin sensitivity occurs independently of weight loss.44 These physiological versus pathophysiological effects via common signaling pathways reflect the tissue specificity, heterogeneity, and complexity of GLP-1 biology.
Improved Metabolic Health: Lifestyle modifications that incorporate regular exercise and a healthful diet may sufficiently treat or prevent MetS and perhaps reduce PCa risk; however, pharmacologic interventions provide additional benefit. GLP1RA target multiple components of MetS, particularly diabetes and obesity (TABLE 1). As incretin mimetics, they augment GSIS by pancreatic beta cells, improving glucose tolerance and reducing plasma glucose. GLP1RA-mediated suppression of food intake and weight loss improve obesity-associated adiposopathy, inflammation, oxidative stress, dyslipidemia, and glucolipotoxicity. Improved endothelial function imparts CV benefits that reduce the risk of atherosclerotic CV disease, coronary artery disease, heart failure, and stroke.45
Enhanced Efficacy of ADT: ADT does not cure PCa; rather, it slows disease progression and allows more time for curative treatments, such as radiotherapy.46 Ironically, one disadvantage of ADT is a heightened risk of diabetes.47 PCa prognosis also worsens, and mortality risk increases.47 DHT enhances GSIS and amplifies the insulinotropic actions of GLP-1 due to increased IR and hyperglycemia.48 Adjuvant treatment with GLP1RA may mitigate ADT-induced impairments in glycemic control. GLP1RA also exert synergistic effects with currently used antiandrogen agents, such as enzalutamide. Although this agent generally improves patient outcomes, it can lose efficacy over time owing to the development of resistance. Use of Ex4 in combination with enzalutamide dramatically suppresses PCa cell growth whereby Ex4 enhances sensitivity to the antiandrogen medication by inhibiting AKT and mTOR activation as well as reducing nuclear AR translocation.49
Antineoplastic Effects: The GLP1R, which is expressed in pancreatic islets, brain, stomach, heart, and kidney, is also prevalent in human PCa tissue. GLP1R messenger RNA is abundantly expressed in androgen-sensitive PCa cell lines but is lower in androgen-independent PCa cell lines.12 Using Ex4 to treat PCa cells overexpressing the GLP1R inhibits cell proliferation, which is prevented by the GLP1R antagonist exendin(9-39)amide and PKA inhibition.50,51 These observations suggest that the antiproliferative effect of Ex4 is due to GLP1RA activation and inhibition of extracellular signal-related kinase (ERK)/MAPK. This phenomenon is greatest in androgen-sensitive PCa cell lines, likely attributable to increased GLP1R expression.12 GLP1R activation also inhibits PCa cell proliferation by halting cell cycle progression.50,51 Furthermore, Ex4 sensitizes PCa to radiotherapy.10 As with enzalutamide, resistance can diminish the efficacy of the antineoplastic agent docetaxel, a microtubule assembly inhibitor and first-line chemotherapy agent for metastatic PCa. However, the efficacy of docetaxel is enhanced when it is used in combination with liraglutide.52 Combination treatment synergistically reduces levels of phosphorylated ERK1/2 and AKT to arrest the cell cycle, inhibit cell proliferation, induce apoptosis, and decrease PCa cell viability.52 Recent findings also suggest that GLP1RA are effective in advanced-stage metastatic PCa, as they inhibit the PI3K/AKT/mTOR and ERK/MAPK pathways.53 In preclinical mouse studies, Ex4 and the first-line antidiabetic medication metformin have been found to decrease PCa cell proliferation.54
Consensus is lacking on whether DPP-IVi are helpful or deleterious in PCa. Some evidence indicates reduced risk and increased survivability in PCa patients using DPP-IVi, particularly compared with other cancer types (e.g., breast, pancreatic).55 CD26, a glycoprotein with intrinsic DPP-IV activity, is implicated in cancer progression and tumor malignancy primarily via enhanced T-cell effector functions.56,57 Elevated CD26 levels are heavily correlated with poor PCa prognosis, and DPP-IV inhibition improves overall survival with no increased risk of metastasis.55,57,58 However, other lines of evidence indicate that DPP-IVi may potentiate PCa tumor growth.59 CD26/DPP-IV inhibition enhances PCa invasion and metastasis.60,61 With ADT, DPP-IVi increase resistance to castration.59 In preclinical studies, sitagliptin-treated mice displayed increased tumor size versus controls.59 ADT resistance is likely due to upregulation of insulin-like growth factor 1, which functionally increases resistance to ADT.62 Furthermore, a survival benefit of DPP-IVi was not observed in diabetic patients with advanced PCa.63 More studies are necessary to fully determine the efficacy of DPP-IVi in PCa.
Challenges With Incretin-Based Pharmacotherapy in PCa
FDA approved since 2005, incretin-based agents are now routinely employed as monotherapy or in combination treatment regimens. Incretin-dependent therapies exert exceptional antihyperglycemic effects partly by potentiating GSIS and improving insulin sensitivity. Therapeutic outcomes are further improved by other metabolic and CV benefits, including the robust weight loss–promoting activity of GLP1RA.53 Despite relatively favorable safety profiles, multiple and potentially serious side effects as well as contraindications are associated with short-term and long-term use of incretin-based drugs. The risk of hypoglycemia is reduced when incretin-based drugs are combined with insulin sensitizers, such as metformin or thiazolidinediones.4,64,65 However, the risk is increased when incretin-based agents are administered with insulin and sulfonylureas, which stimulate pancreatic insulin secretion regardless of serum glucose level.66,67 In this case, a reduction in sulfonylurea dose is recommended.67
GLP1RA: Short-term side effects of GLP1RA range from mild to severe and are primarily GI, including nausea, vomiting, loss of appetite, indigestion, heartburn, abdominal pain, constipation, and diarrhea.68,69 Additional side effects are injection-site pain (e.g., rash, itching, erythema), dehydration, headache, dizziness, and nasopharyngitis.68 These adverse effects are dose-dependent and usually wane over time, so discontinuation of the drug is not typically required.70 Long-term side effects include increased risk of pancreatitis, pancreatic cancer, thyroid carcinoma, neuroendocrine tumors, and acute kidney injury.68 Signs of acute pancreatitis include persistent, severe abdominal pain that may also be lumbar; medical attention should be sought.70 Accordingly, GLP1RA are contraindicated in patients with pancreatitis.68 Dosage adjustment in patients with renal failure is recommended only when creatinine clearance is severely impaired.71
DPP-IVi: Common GI side effects of DPP-IVi include nausea, abdominal pain, and diarrhea. DPP-IVi are also linked to inflammatory bowel disease.72 Additional side effects are upper respiratory tract infection, nasopharyngitis, headache, arthralgia, and urinary tract infection.73,74 Serious adverse events include anaphylaxis, angioedema, and Stevens-Johnson syndrome.73 DPP-IVi are contraindicated in patients with hypersensitivity reactions to drug formulations, diabetic ketoacidosis, type 1 diabetes, and pancreatitis.73 The risk of heart failure has not yet been clarified.75,76 In patients with renal impairment, dosage adjustments are necessary to minimize the potential for hypoglycemia.73
The Pharmacist’s Role
As frontline healthcare providers, pharmacists are well poised to educate patients about PCa prevention, including the benefits of regular, vigorous exercise and consumption of a diet high in fruits, vegetables, and plant-derived beverages, which contain risk-reducing antioxidants.77 Alternatively, diets high in animal and saturated fat as well as excessively high in dairy calcium (>2,000 mg/day) are implicated in disease etiology.78,79
Pharmacists can also counsel patients on how to manage incretin agent–induced side effects, including when medical attention is advised. To mitigate adverse GI symptoms, pharmacists can recommend gradual dose escalation, dietary modifications (e.g., reducing meal size, stopping when full), increased fiber and water intake, use of a stool softener, and light exercise.70 Cautiously used proton-pump inhibitors, histamine H2 receptor blockers, and antiemetics are additional options, as is switching to an alternative agent.70 GLP1RA are slightly less tolerated than DPP-IVi, largely owing to increased nausea and the subcutaneous route of administration (excluding oral semaglutide [Rybelsus]), but they provide superior glycemic control and weight loss in T2D patients.80 Despite inconclusive evidence regarding their antineoplastic actions, DPP-IVi may be preferable when weight is not a concern, oral administration is desirable, or a GLP1RA is intolerable.80 DDP-IVi are less expensive than GLP1RA, but both medications are more costly than non–incretin-based drugs. The precise place of incretin-based agents in PCa pharmacotherapy remains to be elucidated.
REFERENCES
1. Watanabe JH, Kwon J, Nan B, Reikes A. Trends in glucagon-like peptide 1 receptor agonist use, 2014 to 2022. J Am Pharm Assoc (2003). 2024;64(1):133-138.
2. Vaduganathan M, Singh A, Sharma A, et al. Contemporary trends in prescription of dipeptidyl peptidase-4 inhibitors in the context of US Food and Drug Administration warnings of heart failure risk. Am J Cardiol. 2020;125(10):1577-1581.
3. Drucker DJ. GLP-1 physiology informs the pharmacotherapy of obesity. Mol Metab. 2022;57:101351.
4. Gilbert MP, Pratley RE. GLP-1 analogs and DPP-4 inhibitors in type 2 diabetes therapy: review of head-to-head clinical trials. Front Endocrinol (Lausanne). 2020;11:178.
5. Skriver C, Friis S, Knudsen LB, et al. Potential preventive properties of GLP-1 receptor agonists against prostate cancer: a nationwide cohort study. Diabetologia. 2023;66(11):2007-2016.
6. Sieve V. Prostate cancer. In: Salem Press Encyclopedia of Health. Research Starters [database]; 2022.
7. Rawla P. Epidemiology of prostate cancer. World J Oncol. 2019;10(2):63-89.
8. National Cancer Institute Surveillance, Epidemiology, and End Results Program. Cancer stat facts: prostate cancer. https://seer.cancer.gov/statfacts/html/prost.html. Accessed February 28, 2024.
9. Wells KV, Krackeler ML, Jathal MK, et al. Prostate cancer and bone: clinical presentation and molecular mechanisms. Endocr Relat Cancer. 2023;30(9):e220360.
10. He W, Li J. Exendin-4 enhances radiation response of prostate cancer. Prostate. 2018;78(15):1125-1133.
11. Stein MS, Kalff V, Williams SG, et al. The GLP-1 receptor is expressed in vivo by human metastatic prostate cancer. Endocr Oncol. 2024;4(1):e230015.
12. Nomiyama T, Kawanami T, Irie S, et al. Exendin-4, a GLP-1 receptor agonist, attenuates prostate cancer growth. Diabetes. 2014;63(11):3891-3905.
13. Davey RA, Grossmann M. Androgen receptor structure, function and biology: from bench to bedside. Clin Biochem Rev. 2016;37(1):3-15.
14. Pungsrinont T, Kallenbach J, Baniahmad A. Role of PI3K-AKT-mTOR pathway as a pro-survival signaling and resistance-mediating mechanism to therapy of prostate cancer. Int J Mol Sci. 2021;22(20):11088.
15. Audet-Walsh É, Dufour CR, Yee T, et al. Nuclear mTOR acts as a transcriptional integrator of the androgen signaling pathway in prostate cancer. Genes Dev. 2017;31(12):1228-1242.
16. Mamouni K, Kallifatidis G, Lokeshwar BL. Targeting mitochondrial metabolism in prostate cancer with triterpenoids. Int J Mol Sci. 2021;22(5):2466.
17. Shorning BY, Dass MS, Smalley MJ, Pearson HB. The PI3K-AKT-mTOR pathway and prostate cancer: at the crossroads of AR, MAPK, and WNT signaling. Int J Mol Sci. 2020;21(12):4507.
18. Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene. 2013;32(49):5501-5511.
19. Higano CS, Crawford ED. New and emerging agents for the treatment of castration-resistant prostate cancer. Urol Oncol. 2011;29(Suppl 6):s1-s8.
20. da Silva HB, Amaral EP, Nolasco EL, et al. Dissecting major signaling pathways throughout the development of prostate cancer. Prostate Cancer. 2013;2013:920612.
21. Zhao G, Forn-Cuní G, Scheers M, et al. Simultaneous targeting of AMPK and mTOR is a novel therapeutic strategy against prostate cancer. Cancer Lett. 2024;587:216657.
22. Li D, Xu W, Chang Y, et al. Advances in landscape and related therapeutic targets of the prostate tumor microenvironment. Acta Biochim Biophys Sin (Shanghai). 2023;55(6):956-973.
23. Dallmeier D, Larson MG, Vasan RS, et al. Metabolic syndrome and inflammatory biomarkers: a community-based cross-sectional study at the Framingham Heart Study. Diabetol Metab Syndr. 2012;4(1):28.
24. Fontana L, Eagon JC, Trujillo ME, et al. Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes. 2007;56(4):1010-1013.
25. Blüher M, Fasshauer M, Tönjes A, et al. Association of interleukin-6, C-reactive protein, interleukin-10 and adiponectin plasma concentrations with measures of obesity, insulin sensitivity and glucose metabolism. Exp Clin Endocrinol Diabetes. 2005;113(9):534-537.
26. Saydah S, Ballard-Barbash R, Potischman N. Association of metabolic syndrome with insulin-like growth factors among adults in the US. Cancer Causes Control. 2009;20(8):1309-1316.
27. Sousa AP, Costa R, Alves MG, et al. The impact of metabolic syndrome and type 2 diabetes mellitus on prostate cancer. Front Cell Dev Biol. 2022;10:843458.
28. Di Sebastiano KM, Pinthus JH, Duivenvoorden WCM, et al. Elevated C-peptides, abdominal obesity, and abnormal adipokine profile are associated with higher Gleason scores in prostate cancer. Prostate. 2017;77(2):211-221.
29. Lavalette C, Trétarre B, Rebillard X, et al. Abdominal obesity and prostate cancer risk: epidemiological evidence from the EPICAP study. Oncotarget. 2018;9(77):34485-34494.
30. Laukkanen JA, Laaksonen DE, Niskanen L, et al. Metabolic syndrome and the risk of prostate cancer in Finnish men: a population-based study. Cancer Epidemiol Biomarkers Prev. 2004;13(10):1646-1650.
31. Feng X, Song M, Preston MA, et al. The association of diabetes with risk of prostate cancer defined by clinical and molecular features. Br J Cancer. 2020;123(4):657-665.
32. Helsen C, Van den Broeck T, Voet A, et al. Androgen receptor antagonists for prostate cancer therapy. Endocr Relat Cancer. 2014;21(4):T105-T118.
33. Narayan V, Ross AE, Parikh RB, et al. How to treat prostate cancer with androgen deprivation and minimize cardiovascular risk: a therapeutic tightrope. JACC CardioOncol. 2021;3(5):737-741.
34. Banerjee PP, Banerjee S, Brown TR, Zirkin BR. Androgen action in prostate function and disease. Am J Clin Exp Urol. 2018;6(2):62-77.
35. Freedland SJ, Abrahamsson PA. Androgen deprivation therapy and side effects: are GnRH antagonists safer? Asian J Androl. 2021;23(1):3-10.
36. Rowlands J, Heng J, Newsholme P, Carlessi R. Pleiotropic effects of GLP-1 and analogs on cell signaling, metabolism, and function. Front Endocrinol (Lausanne). 2018;9:672.
37. Aroor AR, Sowers JR, Jia G, DeMarco VG. Pleiotropic effects of the dipeptidylpeptidase-4 inhibitors on the cardiovascular system. Am J Physiol Heart Circ Physiol. 2014;307(4):H477-H492.
38. Ansari HUH, Qazi SU, Sajid F, et al. Efficacy and safety of glucagon-like peptide-1 receptor agonists on body weight and cardiometabolic parameters in individuals with obesity and without diabetes: a systematic review and meta-analysis. Endocr Pract. 2024;30(2):160-171.
39. Lu S, Yin H, Yu OHY, Azoulay L. Incretin-based drugs and the incidence of prostate cancer among patients with type 2 diabetes. Epidemiology. 2022;33(4):563-571.
40. Burmeister MA, Brown JD, Ayala JE, et al. The glucagon-like peptide-1 receptor in the ventromedial hypothalamus reduces short-term food intake in male mice by regulating nutrient sensor activity. Am J Physiol Endocrinol Metab. 2017;313(6):e651-e662.
41. Portha B, Tourrel-Cuzin C, Movassat J. Activation of the GLP-1 receptor signalling pathway: a relevant strategy to repair a deficient beta-cell mass. Exp Diabetes Res. 2011;2011:376509.
42. Yang M, Wang J, Wu S, et al. Duodenal GLP-1 signaling regulates hepatic glucose production through a PKC-d-dependent neurocircuitry. Cell Death Dis. 2017;8(2):e2609.
43. Burmeister MA, Ferre T, Ayala JE, et al. Acute activation of central GLP-1 receptors enhances hepatic insulin action and insulin secretion in high-fat-fed, insulin resistant mice. Am J Physiol Endocrinol Metab. 2012;302(3):e334-e343.
44. Mashayekhi M, Nian H, Mayfield D, et al. Weight loss-independent effect of liraglutide on insulin sensitivity in idividuals with obesity and prediabetes. Diabetes. 2024;73(1):38-50.
45. Wang JY, Wang QW, Yang XY, et al. GLP-1 receptor agonists for the treatment of obesity: role as a promising approach. Front Endocrinol (Lausanne). 2023;14:1085799.
46. Wasim S, Park J, Nam S, Kim J. Review of current treatment intensification strategies for prostate cancer patients. Cancers (Basel). 2023;15(23):5615.
47. Lee J, Giovannucci E, Jeon JY. Diabetes and mortality in patients with prostate cancer: a meta-analysis. Springerplus. 2016;5(1):1548.
48. Xu W, Qadir MMF, Nasteska D, et al. Architecture of androgen receptor pathways amplifying glucagon-like peptide-1 insulinotropic action in male pancreatic b cells. Cell Rep. 2023;42(5):112529.
49. Wenjing H, Shao Y, Yu Y, et al. Exendin-4 enhances the sensitivity of prostate cancer to enzalutamide by targeting Akt activation. Prostate. 2020;80(5):367-375.
50. Shigeoka T, Nomiyama T, Kawanami T, et al. Activation of overexpressed glucagon-like peptide-1 receptor attenuates prostate cancer growth by inhibiting cell cycle progression. J Diabetes Investig. 2020;11(5):1137-1149.
51. Knura M, Garczorz W, Borek A, et al. The influence of anti-diabetic drugs on prostate cancer. Cancers (Basel). 2021;13(8):1827.
52. Eftekhari S, Montazeri H, Tarighi P. Synergistic anti-tumor effects of liraglutide, a glucagon-like peptide-1 receptor agonist, along with docetaxel on LNCaP prostate cancer cell line. Eur J Pharmacol. 2020;878:173102.
53. Zhao X, Wang M, Wen Z, et al. GLP-1 receptor agonists: beyond their pancreatic effects. Front Endocrinol (Lausanne). 2021;12:721135.
54. Tsutsumi Y, Nomiyama T, Kawanami T, et al. Combined treatment with exendin-4 and metformin attenuates prostate cancer growth. PLoS One. 2015;10(10):e0139709.
55. Shah C, Hong YR, Bishnoi R, et al. Impact of DPP4 inhibitors in survival of patients with prostate, pancreas, and breast cancer. Front Oncol. 2020;10:405.
56. Ohnuma K, Hosono O, Dang NH, Morimoto C. Dipeptidyl peptidase in autoimmune pathophysiology. Adv Clin Chem. 2011;53:51-84.
57. Lu Z, Qi L, Bo XJ, et al. Expression of CD26 and CXCR4 in prostate carcinoma and its relationship with clinical parameters. J Res Med Sci. 2013;18(8):647-652.
58. Rathmann W, Kostev K. Association of dipeptidyl peptidase 4 inhibitors with risk of metastases in patients with type 2 diabetes and breast, prostate or digestive system cancer. J Diabetes Complications. 2017;31(4):687-692.
59. Russo JW, Gao C, Bhasin SS, et al. Downregulation of dipeptidyl peptidase 4 accelerates progression to castration-resistant prostate cancer. Cancer Res. 2018;78(22):6354-6362.
60. Wesley UV, McGroarty M, Homoyouni A. Dipeptidyl peptidase inhibits malignant phenotype of prostate cancer cells by blocking basic fibroblast growth factor signaling pathway. Cancer Res. 2005;65(4):1325-1334.
61. Sun YX, Pedersen EA, Shiozawa Y, et al. CD26/dipeptidyl peptidase IV regulates prostate cancer metastasis by degrading SDF-1/CXCL12. Clin Exp Metastasis. 2008;25(7):765-776.
62. Liu G, Zhu M, Zhang M, Pan F. Emerging role of IGF-1 in prostate cancer: a promising biomarker and therapeutic target. Cancers (Basel). 2023;15(4):1287.
63. Pan K, Skelton WP, Elzeneini M, et al. A multi-center retrospective analysis examining the effect of dipeptidyl peptidase-4 inhibitors on progression-free survival in patients with prostate cancer. Cureus. 2021;13(4):e14712.
64. Ahrén B. Novel combination treatment of type 2 diabetes DPP-4 inhibition + metformin. Vasc Health Risk Manag. 2008;4(2):383-394.
65. Rosenstock J, Brazg R, Andryuk PJ, et al. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther. 2006;28(10):1556-1568.
66. de Heer J, Holst JJ. Sulfonylurea compounds uncouple the glucose dependence of the insulinotropic effect of glucagon-like peptide 1. Diabetes. 2007;56(2):438-443.
67. Salvo F, Moore N, Arnaud M, et al. Addition of dipeptidyl peptidase-4 inhibitors to sulphonylureas and risk of hypoglycaemia: systematic review and meta-analysis. BMJ. 2016;353:i2231.
68. Filippatos TD, Panagiotopoulou TV, Elisaf MS. Adverse effects of GLP-1 receptor agonists. Rev Diabet Stud. 2014;11(3-4):202-230.
69. Liu BD, Udemba SC, Liang K, et al. Shorter-acting glucagon-like peptide-1 receptor agonists are associated with increased development of gastro-oesophageal reflux disease and its complications in patients with type 2 diabetes mellitus: a population-level retrospective matched cohort study. Gut. 2024;73(2):246-254.
70. Wharton S, Davies M, Dicker D, et al. Managing the gastrointestinal side effects of GLP-1 receptor agonists in obesity: recommendations for clinical practice. Postgrad Med. 2022;134(1):14-19.
71. Yu JH, Park SY, Lee DY, et al. GLP-1 receptor agonists in diabetic kidney disease: current evidence and future directions. Kidney Res Clin Pract. 2022;41(2):136-149.
72. Abrahami D, Douros A, Yin H, et al. Dipeptidyl peptidase-4 inhibitors and incidence of inflammatory bowel disease among patients with type 2 diabetes: population based cohort study. BMJ. 2018;360:k872.
73. Pathak R, Bridgeman MB. Dipeptidyl peptidase-4 (DPP-4) inhibitors in the management of diabetes. P T. 2010;35(9):509-513.
74. Men P, He N, Song C, Zhai S. Dipeptidyl peptidase-4 inhibitors and risk of arthralgia: a systematic review and meta-analysis. Diabetes Metab. 2017;43(6):493-500.
75. Packer M. Worsening heart failure during the use of DPP-4 inhibitors: pathophysiological mechanisms, clinical risks, and potential influence of concomitant antidiabetic medications. JACC Heart Fail. 2018;6(6):445-451.
76. Li L, Li S, Deng K, et al. Dipeptidyl peptidase-4 inhibitors and risk of heart failure in type 2 diabetes: systematic review and meta-analysis of randomised and observational studies. BMJ. 2016;352:i610.
77. Vance TM, Su J, Fontham ETH, et al. Dietary antioxidants and prostate cancer: a review. Nutr Cancer. 2013;65(6):793-801.
78. Di Sebastiano KM, Mourtzakis M. The role of dietary fat throughout the prostate cancer trajectory. Nutrients. 2014;6(12):6095-6109.
79. Park Y, Mitrou PN, Kipnis V, et al. Calcium, dairy foods, and risk of incident and fatal prostate cancer: the NIH-AARP Diet and Health Study. Am J Epidemiol. 2007;166(11):1270-1279.
80. Brunton S. GLP-1 receptor agonists vs. DPP-4 inhibitors for type 2 diabetes: is one approach more successful or preferable than the other? Int J Clin Pract. 2014;68(5):557-567.
81. Rizzo M, Nikolic D, Patti AM, et al. GLP-1 receptor agonists and reduction of cardiometabolic risk: potential underlying mechanisms. Biochim Biophys Acta Mol Basis Dis. 2018;1864(9 Pt B):2814-2821.
82. Martins FF, Marinho TS, Cardoso LEM, et al. Semaglutide (GLP-1 receptor agonist) stimulates browning on subcutaneous fat adipocytes and mitigates inflammation and endoplasmic reticulum stress in visceral fat adipocytes of obese mice. Cell Biochem Funct. 2022;40(8):903-913.
83. Simental-Mendía LE, Sánchez-García A, Linden-Torres E, Simental-Mendía M. Impact of glucagon-like peptide-1 receptor agonists on adiponectin concentrations: a meta-analysis of randomized controlled trials. Br J Clin Pharmacol. 2021;87(11):4140-4149.
84. Simental-Mendía LE, Sánchez-García A, Linden-Torres E, Simental-Mendía M. Effect of glucagon-like peptide-1 receptor agonists on circulating levels of leptin and resistin: a meta-analysis of randomized controlled trials. Diabetes Res Clin Pract. 2021;177:108899.
85. Drucker DJ. The cardiovascular biology of glucagon-like peptide-1. Cell Metab. 2016;24(1):15-30.
86. Stemmer K, Finan B, DiMarchi RD, et al. Insights into incretin-based therapies for treatment of diabetic dyslipidemia. Adv Drug Deliv Rev. 2020;159:34-53.
87. Mulvihill EE, Drucker DJ. Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr Rev. 2014;35(6):992-1019.
88. Ussher JR, Drucker DJ. Glucagon-like peptide 1 receptor agonists: cardiovascular benefits and mechanisms of action. Nat Rev Cardiol. 2023;20(7):463-474.
89. Buteau J, El-Assaad W, Rhodes CJ, et al. Glucagon-like peptide-1 prevents beta cell glucolipotoxicity. Diabetologia. 2004;47(5):806-815.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
To comment on this article, contact rdavidson@uspharmacist.com.



