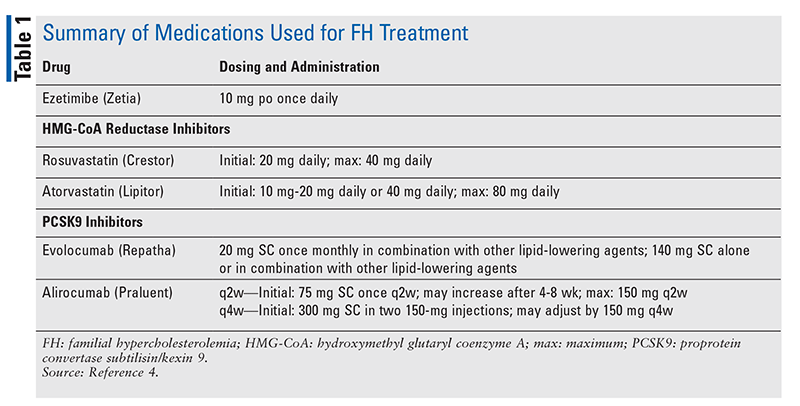
US Pharm. 2020;45(11):31-33.
Elevated serum levels of total cholesterol (TC) and LDL cholesterol (LDL-C) are strong and well-established risk factors for morbidity and/or mortality associated with coronary heart disease (CHD).1 Primary prevention of CHD entails lowering elevated LDL-C levels via dietary interventions and drug therapy, which are effective in most patients. Familial hypercholesterolemia (FH), the most common autosomal-dominant genetic disorder affecting cholesterol metabolism, is usually due to a functional mutation in one of three genes associated with impaired LDL receptor–mediated catabolism, which leads to higher LDL-C in the blood. Patients with heterozygous FH usually respond to standard therapy, whereas the majority of patients with homozygous FH fail to respond. Patients are at higher risk for developing ischemic heart disease at a younger age because of resistance to the effects of diet and medications. In patients whose LDL-C levels cannot be managed by more traditional means, LDL apheresis (a nonsurgical process for removing LDL-C from the blood) has been demonstrated to be effective and safe.1
FH Epidemiology and Mutation
According to the CDC, the National Health and Nutrition Examination Survey for 1999 to 2014 estimated that the prevalence of definite or probable FH in the U.S. adults older than age 20 years was 0.5%.2 Homozygous FH is rare, with an estimated prevalence of approximately one case per 300,000 to 400,000 in the U.S.; heterozygous FH is estimated to occur in about one in 200 to 250 people (approximately 1 million).3 The prevalence is greater in black persons compared with white persons.3
Patients with FH usually have a functional mutation in one of three genes that play a role in cholesterol metabolism: LDL (LDLR), proprotein convertase subtilisin/kexin 9 (PCSK9), and apolipoprotein B (APOB).3 The prevalence of pathogenic mutation in at least one of these genes varies by degree of certainty—either definite or possible FH syndrome.3
FH Diagnosis
Diagnosis of FH depends on whether the patient is heterozygous or homozygous for the gene mutation. In patients with heterozygous FH, diagnosis is made via genetic testing or clinical criteria (fasting lipid profile). The fasting lipid profile usually consists of elevated TC and LDL with normal or low HDL and normal triglyceride (TG) levels unless the patient is obese, has diabetes, or possesses other risk factors that may elevate TG levels.3 Elevated TG levels do not exclude a diagnosis of FH; however, other potential causes of elevated TG must be taken into consideration.3
Genetic testing offers panels that capture major pathogenic LDLRs and analyze the gene sequence.3 There is no widespread agreement as to when genetic testing is indicated.3 In general, testing is ordered when the results may alter the clinical decision; for example, whether to use statins in a woman of childbearing potential with established pathogenic genetic defects contributing to FH. When genetic testing is not available or is deemed unnecessary, the Dutch Lipid Clinic Network criteria—in which points are assigned based on LDL-C, personal history of early atherosclerotic cardiovascular (CV) disease (ASCVD), family history of early ASCVD or high cholesterol in a first-degree relative, and personal and physical-examination findings (tendinous xanthoma or arcus cornealis before age 45 years)—are used.3 A score of 8 points or higher indicates a definitive FH diagnosis.3
The clinical diagnosis of homozygous FH includes untreated LDL-C (500 mg/dL or higher) or treated LDL-C (300 mg/dL or higher) and cutaneous or tendinous xanthoma before age 10 years or elevated LDL-C levels consistent with heterozygous FH in both parents.3
FH Treatment
In both homozygous and heterozygous FH, the treatment goal is intense lowering of LDL-C. All patients with elevated LDL-C should be counseled on lifestyle changes (i.e., dietary modifications, physical activity, and weight loss, if needed) that may reduce LDL-C levels and other CV risks.4 The use of aspirin may also be appropriate for many of these patients because they are at higher risk for a CV event.4 Aspirin is recommended for patients with clinical ASCVD and for men older than age 55 years with multiple CHD risk factors.4
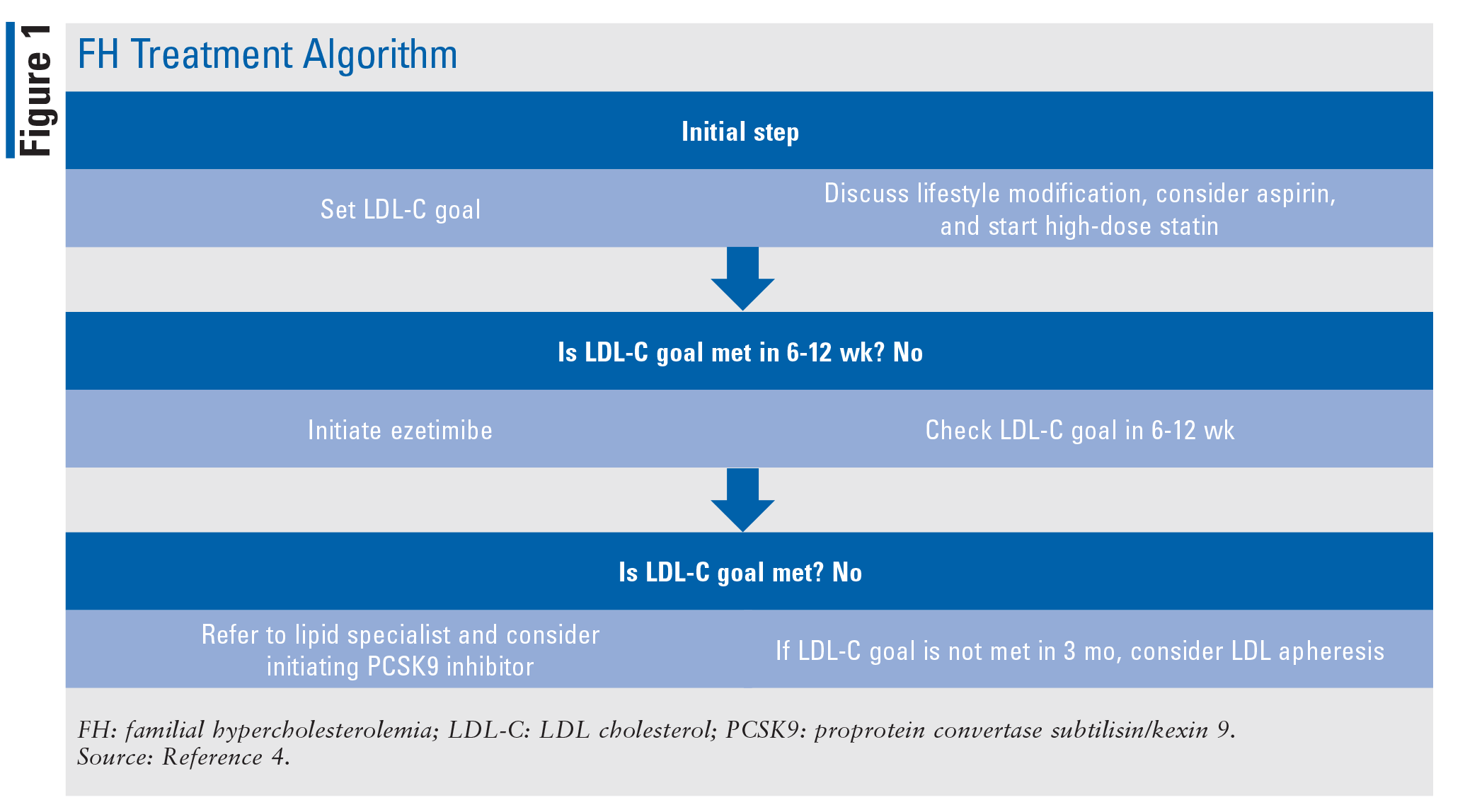
All patients diagnosed with homozygous FH should be referred to a lipid specialist and will benefit from the care of a CV specialist.4 The treatment strategy for lowering LDL-C in homozygous and heterozygous FH patients usually involves the use of a high-dose statin and may include the use of ezetimibe, a PCSK9 inhibitor, or both (TABLE 1). Therapy follows an algorithm that starts with a high-dose statin titrated to the targeted dose (FIGURE 1). Once the patient is stable on a high-dose statin, therapy is reevaluated every 6 to 12 weeks, and adjustments are made to better meet LDL-C goals. Ezetimibe is the next treatment option if high-dose statins are not effective in lowering LDL-C. If it is determined that goals are still not being met with use of ezetimibe plus a high-dose statin, a PCSK9 inhibitor may be added by a lipid specialist. The use of LDL apheresis is the last option for treating FH when LDL-C levels are not being met despite the use of standard therapy.
Liposorber LA-15 System
The Liposorber LA-15 system (FIGURE 2; Kaneka Pharma America LLC) is a device for LDL apheresis. The system includes a tubing set, two blood filters, two blood-storage columns, and a computer that regulates the entire system. By removing LDL-C, very-low-density lipoprotein (VLDL), and lipoprotein(a) from the blood, the Liposorber LA-15 system may reduce the likelihood that the patient develops serious diseases of the heart and blood vessels.5

Several studies have demonstrated the efficacy of LDL apheresis. One randomized, controlled, multicenter trial evaluated LDL-C and lipoprotein(a) removal via the Liposorber LA-15 system in patients with an inadequate response to diet and maximal drug therapy. Of the 64 patients, 54 had heterozygous FH (45 assigned to LDL apheresis treatment and nine controls) and 10 had homozygous FH (all assigned to treatment).6 Treatment was administered at intervals of 7 to 14 days.6 The mean acute reduction in LDL cholesterol was 76% in heterozygous FH patients and 81% in homozygous FH patients.6 Time-averaged levels of LDL-C were reduced 41% (243 mg/dL-143 mg/dL) in heterozygous FH patients and 53% (447 mg/dL-210 mg/dL) in homozygous FH patients.6 It was concluded that the Liposorber LA-15 system is an important therapeutic option for FH patients who have an inadequate response to diet and drug therapy.6
Conclusion
The Liposorber LA-15 system is an LDL-apheresis device that selectively removes LDL-C, VLDL, and lipoprotein(a). The system has demonstrated effectiveness for treating FH patients with elevated TC and LDL-C who do not respond well to standard therapy. See www.fda.gov/medical-devices/recently-approved-devices/liposorberr-la-15-system-h170002 on the FDA’s website for more information.
REFERENCES
1. Agishi T, Wood W, Gordon B. LDL apheresis using the liposorber® LA-15 system in coronary and peripheral vascular disease associated with severe hypercholesterolemia. Curr Ther Res. 1994;55(8):879-904.
2. Bucholz EM, Rodday AM, Kolor K, et al. Familial hypercholesterolemia is common and undertreated in the United States. https://blogs.cdc.gov/genomics/2018/03/26/familial-hypercholesterolemia. Accessed May 22, 2020.
3. Rosenson RS, Durrington P. Familial hypercholesterolemia in adults: overview. UpToDate. Waltham, MA: UpToDate; 2020.
4. Rosenson RS, Durrington P. Familial hypercholesterolemia in adults: treatment. UpToDate. Waltham, MA: UpToDate; 2020.
5. A patient guide to the Liposorber® LA-15 system. www.accessdata.fda.gov/cdrh_docs/pdf17/H170002C.pdf. Accessed January 12, 2020.
6. Gordon BR, Kelsey SF, Bilheimer DW, et al. Treatment of refractory familial hypercholesterolemia by low-density lipoprotein apheresis using an automated dextran sulfate cellulose adsorption system. The Liposorber Study Group. Am J Cardiol. 1992;70(11):1010-1016.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
To comment on this article, contact rdavidson@uspharmacist.com.


