US Pharm. 2011;36(10):39-52.
New molecular entities (NMEs), as defined by the FDA, are new drug products containing as their active ingredient a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in 2010–2011 (TABLE 1) detail the basic clinical and pharmacologic profile of each new drug, as well as key precautions and warnings. Also included for each drug is a brief summary of selected pharmacokinetic, adverse-reaction, drug-interaction, and dosing data submitted to the FDA in support of the manufacturer’s New Drug Application. This review is intended to be objective rather than evaluative in content. The information for each NME was obtained primarily from sources published prior to FDA approval. Experience clearly shows that many aspects of a new drug’s therapeutic profile are not detected in premarketing studies and emerge after the drug is used in large numbers of patients. Studies have clearly demonstrated the appearance of “new” adverse reactions for many NMEs within 2 to 3 years of first becoming available. Some of these drugs may eventually acquire at least one black box warning for serious adverse reactions or are withdrawn from the market for safety reasons not recognized at the time of approval. Hence, while this review offers a starting point for learning about new drugs, it is essential that practitioners be aware of changes in a drug’s therapeutic profile as reported by their own patients and in the pharmaceutical literature.
Azilsartan (Edarbi, Takeda Pharmaceuticals America)
Indication and Clinical Profile1,2: Azilsartan is the newest angiotensin II receptor blocker (ARB) indicated for the treatment of hypertension, alone or in combination with other antihypertensive agents. Hypertension, defined as systolic/diastolic blood pressure (BP) of >140 mmHg/90 mmHg, affects approximately 75 million Americans and is associated with increased risk of stroke, heart failure, heart attack, renal failure (RF), and death. Drugs that block the renin-angiotensin system, including ARBs, ACE inhibitors, and renin inhibitors, have become standard treatments for hypertension.
The efficacy of azilsartan was demonstrated by seven phase III clinical trials (placebo-controlled and active comparator-controlled) involving more than 5,900 patients. These trials showed that azilsartan was statistically superior to placebo and active comparators (olmesartan and valsartan) in the reduction of BP measurements at endpoint.
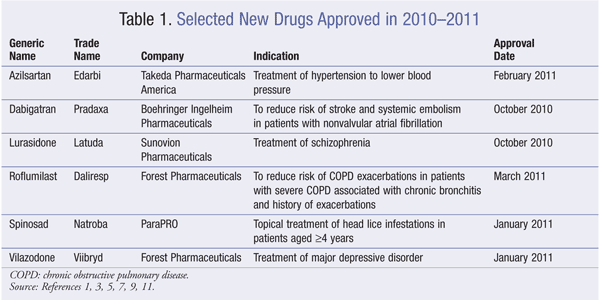
Pharmacology and Pharmacokinetics1,2: Azilsartan is a benzimidazole ester (FIGURE 1) that inhibits the vasoconstrictor and aldosterone-secreting effects of angiotensin II by selectively blocking the binding of angiotensin II to the angiotensin type 1 (AT1) receptor in many tissues, such as vascular smooth muscle and the adrenal gland. The angiotensin type 2 (AT2) receptor is also found in many tissues, but it is not known to be associated with cardiovascular homeostasis. Azilsartan has a >10,000-fold greater affinity for the AT1 receptor than for the AT2 receptor. Blockade of the angiotensin II receptor inhibits the negative regulatory feedback of angiotensin II on renin secretion, but the resulting increased plasma renin activity and angiotensin II circulating levels do not overcome the effect of azilsartan on BP.
Azilsartan medoxomil is rapidly hydrolyzed to azilsartan, the active metabolite, in the gastrointestinal (GI) tract during absorption. The estimated absolute bioavailability of azilsartan is approximately 60% and is not affected by food. Azilsartan is metabolized by O-dealkylation by CYP2C9 (metabolite M-II) and decarboxylation (metabolite M-I). M-I and M-II do not contribute to the drug’s pharmacologic activity. Azilsartan is eliminated in the urine and feces, mainly as metabolites.
Adverse Reactions and Drug Interactions1,2: Adverse reactions to azilsartan in clinical trials were similar to those reported by subjects taking placebo. The most common adverse reactions were dizziness, increased blood creatine phosphokinase, and diarrhea. Azilsartan medoxomil has a black box warning stating that the drug should be avoided during the second and third trimesters of pregnancy because it can cause fetal injury or death (Pregnancy Category D). If a woman becomes pregnant while using azilsartan, the drug should be discontinued as soon as possible. In patients with an activated renin-angiotensin system, as by volume or salt depletion, ARBs can cause excessive hypotension; therefore, volume or salt depletion should be corrected prior to administration. ARBs can cause RF in susceptible patients (e.g., renal artery stenosis), so renal function should be monitored in patients with renal impairment.
Coadministration of nonsteroidal anti-inflammatory drugs (NSAIDs) and ARBs may result in a deterioration in renal function, including possible acute RF, as well as a decreased antihypertensive effect of the ARB.
Dosage and Administration1,2: Azilsartan is supplied as 40-mg and 80-mg tablets. The recommended dose is 80 mg once daily; the 40-mg dose may be used in patients taking high-dose diuretics. No initial dosage adjustment is recommended for elderly patients or in patients with mild-to-severe renal impairment, end-stage renal disease, or mild-to-moderate hepatic dysfunction. Azilsartan has not been studied in patients with severe hepatic impairment. If BP is not controlled with azilsartan alone, additional BP reduction may be achieved through the addition of other antihypertensives.
Dabigatran (Pradaxa, Boehringer Ingelheim Pharmaceuticals)
Indication and Clinical Profile3,4: Dabigatran was approved by the FDA to reduce the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation (AF). It is the first alternative to warfarin to be approved by the FDA. Replacement drugs for warfarin are considered potential blockbusters because of the critical need for them, particularly as the aging population becomes more vulnerable to AF and other conditions that can cause blood clots. AF affects more than 2 million Americans.
The clinical efficacy of dabigatran was demonstrated in the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY) trial, which enrolled 18,113 patients with nonvalvular, persistent, paroxysmal, or permanent AF. Two blinded dosages of dabigatran (110 mg twice daily and 150 mg twice daily) were compared with open-label warfarin dosed to target an international normalized ratio (INR) of 2 to 3. Patients were followed for a median of 2 years. Dabigatran 150 mg twice daily significantly reduced the primary composite endpoint of stroke and systemic embolism compared with both warfarin and dabigatran 110 mg twice daily.
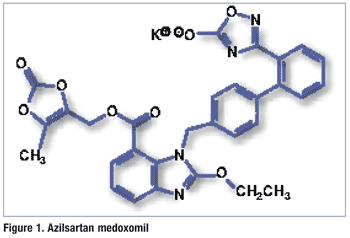
Pharmacology and Pharmacokinetics3,4: Dabigatran is a highly substituted benzimidazole (FIGURE 2) that, along with its acyl glucuronide metabolites, is a competitive direct thrombin inhibitor. Thrombin is a serine protease that converts fibrinogen into fibrin during the coagulation cascade, and its inhibition prevents a thrombus from developing. Both free and clot-bound thrombin and thrombin-induced platelet aggregation are inhibited by the active moieties.
The absolute bioavailability following oral administration of dabigatran etexilate is 3% to 7% and is not significantly affected by food. Bioavailability increases by 75% when the pellets are taken without the capsule shell compared with the intact capsule formulation; therefore, dabigatran capsules should not be broken, chewed, or opened before administration. After oral administration, dabigatran etexilate is converted to dabigatran by esterases. Dabigatran is subject to conjugation, forming four pharmacologically active acyl glucuronides. Dabigatran is eliminated primarily in the urine, with a half-life of 12 to 17 hours.
Adverse Reactions and Drug Interactions3,4: As with other anticoagulants, the most significant adverse reaction associated with dabigatran is bleeding. In the RE-LY trial, the risk of major bleeds was similar between dabigatran 150 mg and adjusted-dose warfarin in all major patient subgroups except age, where dabigatran showed a trend toward a higher incidence of major bleeding. There was a higher rate of major GI bleeds in patients receiving dabigatran 150 mg than in those receiving adjusted-dose warfarin. The risk of myocardial infarction was greater in patients taking dabigatran 150 mg than in those taking warfarin. In trials to date, no QTc-interval prolongation has been observed with dabigatran doses up to 600 mg.
Dabigatran is not a substrate, inhibitor, or inducer of CYP450 enzymes; therefore, interactions with other drugs metabolized by these enzymes are not anticipated. Because dabigatran is a substrate of the efflux transporter P-glycoprotein (Pgp), concurrent use with Pgp inducers can reduce dabigatran exposure and generally should be avoided. Pgp inhibitors do not appear to affect dabigatran’s pharmacokinetics.
Dosage and Administration3,4: Dabigatran is available in 75-mg and 150-mg capsules. The recommended dosage is 150 mg taken orally twice daily, with or without food. The capsules should not be broken, chewed, or emptied, as this may result in increased exposure. For patients with significantly impaired renal function (creatinine clearance [CrCl] 15-30 mL/min), the recommended dosage is 75 mg twice daily. No dosing recommendations are available for patients with CrCl <15 mL/min or for those receiving dialysis. When a patient is being switched from warfarin to dabigatran, warfarin should first be discontinued and dabigatran started only after the INR is <2.0. See the prescribing information for dabigatran for recommendations for converting to or from other oral or parenteral anticoagulants.
Lurasidone (Latuda, Sunovion Pharmaceuticals)
Indication and Clinical Profile5,6: Lurasidone is the newest atypical antipsychotic (AA) agent approved for the treatment of schizophrenia in adults. Annually, schizophrenia affects about 1% of the U.S. population aged >18 years. The disorder is characterized by three symptom types: positive (abnormal behavior), negative (inability to perform daily activities), and cognitive (inability to make decisions).
The development of lurasidone involved more than 40 clinical trials in about 2,700 adults. Efficacy was established by four 6-week placebo-controlled clinical studies in which lurasidone demonstrated significantly greater improvement versus placebo on the primary efficacy measures (Positive and Negative Syndrome Scale [PANSS] total score and Brief Psychiatric Rating Scale derived from PANSS).
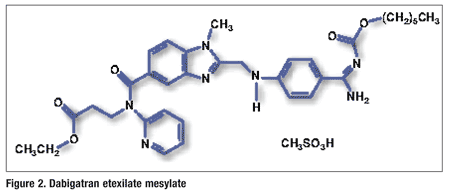
Pharmacology and Pharmacokinetics5,6: Lurasidone is an isoindole derivative (FIGURE 3) that has a unique receptor-binding profile with high affinity for dopamine-2, serotonin-2A, serotonin-7, serotonin-1A, and noradrenaline-2C receptors. These receptors are known to improve cognitive capabilities upon effective regulation. The drug has limited affinity for histamine-1 and acetylcholine-M1 receptors and is a partial agonist for the serotonin 5-hydroxytryptamine (5HT)1A receptor. Lurasidone enhances the functions of muscarinic acetylcholine receptors, which are known to aid in memory and learning functions.
Lurasidone is 9% to 19% absorbed, producing peak serum concentrations in 1 to 3 hours. Administration with food increases both Cmax and AUC. The drug is metabolized mainly via CYP3A4 by pathways including oxidative N-dealkylation, hydroxylation of norbornane ring, and S-oxidation. Lurasidone is metabolized into two active metabolites (ID-14283 and ID-14326) and two major nonactive metabolites (ID-20219 and ID-20220). The drug is primarily cleared in the feces (80%) as metabolites.
Adverse Reactions and Drug Interactions5,6: The most common adverse reactions in clinical trials were drowsiness; akathisia; feelings of restlessness; nausea; movement abnormalities such as tremors, slow movement, or muscle stiffness (parkinsonism); and agitation. All AAs carry a black box warning alerting prescribers to an increased risk of death associated with off-label use to treat behavioral problems in older people with dementia-related psychosis. Lurasidone, like other AAs, comes with warnings for cerebrovascular adverse effects including stroke, neuroleptic malignant syndrome, tardive dyskinesias, orthostatic hypotension, hematologic abnormalities, hyperprolactinemia, seizures, cognitive impairment, suicide, and metabolic abnormalities. Lurasidone is a Pregnancy Category B drug, and use while breastfeeding should be considered only if the potential benefit justifies the potential risk to the child.
Lurasidone is not recommended for use in combination with strong CYP3A4 inhibitors or inducers. Dosage adjustment is recommended when it is used with moderate CYP3A4 inhibitors. Lurasidone is not known to interact with other CYP isozymes, Pgp, or oral contraceptives.
Dosage and Administration5,6: Lurasidone is available in 40-mg and 80-mg tablets. The recommended starting dose is 40 mg once daily, with a maximum dose of 80 mg/day. Higher doses provide no additional benefit and increase the incidence of certain adverse reactions. The dosage should not exceed 40 mg/day in patients with moderate-to-severe renal or hepatic impairment or in patients taking a moderate CYP3A4 inhibitor. Patients taking strong CYP3A4 inhibitors or inducers should not take lurasidone.
Roflumilast (Daliresp, Forest Pharmaceuticals)
Indication and Clinical Profile7,8: Roflumilast is a novel orally active phosphodiesterase 4 (PDE-4) inhibitor approved for the treatment of moderate-to-severe chronic obstructive pulmonary disease (COPD). COPD affects approximately 4% to 10% of the U.S. population and is the fourth leading cause of death. Inflammation appears to be the primary cause of the progressive airflow limitation seen in COPD, resulting in the characteristic symptoms of breathlessness, chronic cough, and excessive phlegm. COPD exacerbations may last up to several weeks, be associated with severe anxiety, and result in lung-function decline and increased risk of death. The anti-inflammatory agents and bronchodilators available for COPD treatment do not reduce the likelihood of disease progression.
The efficacy of roflumilast was demonstrated in two phase III clinical studies involving more than 1,500 patients aged >40 years. Patients had a history of COPD associated with chronic bronchitis and had experienced COPD exacerbation in the 12 months before treatment initiation. In these trials, roflumilast improved markers of lung function, including forced expiratory volume in 1 second and forced vital capacity, and reduced the incidence of COPD exacerbations, particularly in patients with more severe disease. These benefits occurred with monotherapy and combined therapy.
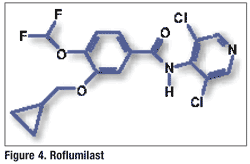
Pharmacology and Pharmacokinetics7,8: Roflumilast is a substituted benzamide (FIGURE 4) that, along with its active metabolite (roflumilast N-oxide), selectively inhibits PDE-4. This inhibition results in an increase in intracellular cyclic adenosine monophosphate in respiratory tissue. The exact mechanism of roflumilast in COPD is unknown, but it is theorized that the drug may express multiple anti-inflammatory effects by inhibiting PDE-4 expressed on immune and proinflammatory cells. Roflumilast has been shown to significantly reduce sputum neutrophils and eosinophils in COPD patients. However, there is no evidence that roflumilast has a bronchodilation effect.
The absolute bioavailability of roflumilast is approximately 80%. Food has no effect on total drug absorption, but it delays time to maximum concentration (Tmax) by 1 hour and reduces Cmax by approximately 40%; Tmax and Cmax of roflumilast N-oxide are unaffected, however. Roflumilast is extensively metabolized via cytochrome oxidation (CYP1A2 and CYP3A4) and conjugation. The major cytochrome metabolite—the N-oxide—and the parent drug account for the majority (87.5%) of total drug in plasma. While roflumilast is three times more potent than roflumilast N-oxide as a PDE-4 inhibitor, plasma levels of roflumilast N-oxide are approximately 10-fold greater than roflumilast levels. The plasma half-lives of roflumilast and its N-oxide metabolite are approximately 17 and 30 hours, respectively. Roflumilast is cleared primarily by the kidney, mainly as metabolites including conjugates.
Adverse Reactions and Drug Interactions7,8: The most common adverse reactions in clinical trials were diarrhea, nausea, vomiting, abdominal pain, weight loss, and headache. These reactions occurred mainly within the first weeks of therapy and mostly resolved on continued treatment. Psychiatric adverse events such as suicide (3 suicides and 2 suicide attempts), insomnia, anxiety, and depression also were noted. Adverse cardiovascular events, seen with some other PDE inhibitors, have not been shown to be a concern with roflumilast use of <1 year. Cancer incidence was higher in roflumilast-treated patients: Of 218 cancer or tumor events, 60% occurred in the roflumilast group versus 40% in the placebo group. Roflumilast should not be used by nursing women, as excretion of the drug and its metabolites into human milk is probable. The drug should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus (Pregnancy Category C).
No significant interactions were observed with other drugs likely to be coadministered with roflumilast, including inhaled salbutamol, formoterol, budesonide, oral montelukast, digoxin, theophylline, warfarin, sildenafil, midazolam, and antacids. Use with CYP3A4 inhibitors or dual inhibitors of CYP3A4 and CYP1A2 (erythromycin, ketoconazole, fluvoxamine, enoxacin, cimetidine) increases roflumilast systemic exposure and may result in increased adverse reactions. Use with strong cytochrome enzyme inducers is not recommended.
Dosage and Administration7,8: Roflumilast is supplied as 500-mcg white tablets. The recommended dosage is one 500-mcg tablet per day, with or without food. The drug is contraindicated in moderate-to-severe liver impairment.
Spinosad (Natroba, ParaPRO)
Indication and Clinical Profile9,10: Spinosad topical suspension 0.9% is approved for the treatment of head lice infestation in patients aged >4 years. Head lice, a common problem among U.S. schoolchildren, are spread mainly by direct head-to-head contact with someone who already has head lice. Spinosad provides an alternative to OTC shampoos containing pyrethrin or permethrin and prescription medications including malathion, lindane, and benzyl alcohol lotion.
The safety and effectiveness of spinosad were established in two multicenter, randomized, controlled studies involving 1,038 subjects aged >6 months with head lice infestation. A total of 552 subjects received a 10-minute spinosad treatment; the remaining subjects were treated with permethrin. Subjects treated on day 0 returned for efficacy evaluation on day 7. Spinosad subjects with live lice present on day 7 received a second treatment. Subjects who were lice free on day 7 returned on day 14 for evaluation. Subjects with live lice and those who received a second treatment returned on days 14 and 21. The proportion of subjects who were lice free 14 days after the final application of spinosad was approximately 86%, versus 44% of subjects receiving permethrin.
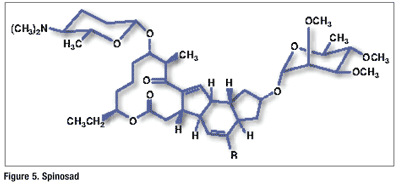
Pharmacology and Pharmacokinetics9,10: Spinosad, which is derived from fermentation of Saccharopolyspora spinosa, is a mixture of spinosyn A and spinosyn D in a ratio of 5 to 1 (FIGURE 5). This natural product kills susceptible species by causing rapid excitation of the insect’s nervous system. Spinosad must be ingested by the insect; therefore, it has little effect on nontarget predatory insects. It is relatively fast acting, with insect death occurring within 1 to 2 days; insect mortality appears to be 100%.
In pharmacokinetic studies, subjects who received a single scalp treatment of spinosad showed no measurable plasma levels of drug. The bioavailability of benzyl alcohol from the product formulation was not determined in these subjects.
Adverse Reactions and Drug Interactions9,10: Common adverse events associated with spinosad therapy include redness or irritation of the eyes and skin. Spinosad is not approved for use in children aged <4 years. It is especially important not to use the product in infants because it contains benzyl alcohol, which has been associated with serious adverse reactions, including death, when applied topically in children aged <6 months. Spinosad is a Pregnancy Category B drug. Because of the lack of systemic absorption, no drug interactions are anticipated with other medications.
Dosage and Administration9,10: Spinosad is supplied as a 0.9%, viscous, slightly opaque, light orange–colored suspension. It should be applied to cover dry scalp and then to dry hair. After 10 minutes, the covered areas should be thoroughly rinsed with warm water. If live lice are seen 7 days after the first treatment, a second treatment should be applied. The drug product is not for oral, ophthalmic, or intravaginal use.
Vilazodone (Viibryd, Forest Pharmaceuticals)
Indication and Clinical Profile11,12: Vilazodone is the newest selective serotonin reuptake inhibitor (SSRI) approved for the treatment of major depressive disorder (MDD). MDD, affecting nearly 19 million people in the U.S., is characterized by depressed mood, loss of interest in usual activities, significant change in weight or appetite, sleep disorders, increased fatigue, impaired concentration, and suicidal thoughts or behavior. The most commonly prescribed drugs for MDD are SSRIs and serotonin-norepinephrine reuptake inhibitors (SNRIs). These agents are preferred because of their relative efficacy and safety and because they are less toxic in overdose than older antidepressants (i.e., tricyclics and monoamine oxidase inhibitors [MAOIs]).
Approval of vilazodone was based on two 8-week, multicenter, randomized, double-blind, placebo-controlled studies in 869 adults with MDD who received either vilazodone titrated over 2 weeks to a dosage of 40 mg or placebo once daily. Vilazodone was superior to placebo in the improvement of depressive symptoms as measured by the mean change from baseline to week 8 in the Montgomery-Asberg Depression Rating Scale total score.
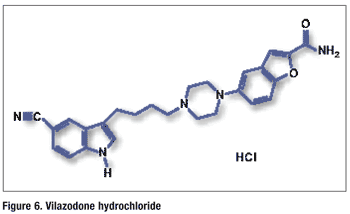
Pharmacology and Pharmacokinetics11,12: Vilazodone is a indole-piperazine (FIGURE 6) that functions as an SSRI and 5-HT1A receptor partial agonist. It has negligible affinity for other serotonin receptors. Vilazodone’s antidepressant effects are thought to be due to enhancement of serotonergic activity in the central nervous system (CNS) through selective inhibition of serotonin reuptake. The contribution of vilazodone’s 5-HT1A receptor partial agonist activity is unknown.
The absolute bioavailability of vilazodone is 72% when the drug is taken with food. Vilazodone is extensively metabolized through CYP and non-CYP pathways, with less than 2% of the dose excreted in the urine and feces as unchanged vilazodone. CYP3A4 is primarily responsible for vilazodone’s metabolism, with minor contributions from CYP2C19 and CYP2D6. Strong inhibitors of CYP3A4 can reduce the metabolism of vilazodone and increase exposure, while CYP3A4 inducers can decrease exposure. Vilazodone does not appear to significantly inhibit or induce the metabolism of other CYP drug substrates (except for CYP2C8).
Adverse Reactions and Drug Interactions11,12: The most frequent adverse reactions reported by patients taking vilazodone in clinical trials were diarrhea, nausea, vomiting, and insomnia. Vilazodone has a rate of sexual side effects and weight gain comparable to placebo. All antidepressants have a black box warning and a patient medication guide describing the increased risk of suicidal thinking and behavior in children, adolescents, and young adults aged 18 to 24 years during initial treatment. Neonates exposed to serotonergic antidepressants late in the third trimester have developed complications requiring prolonged hospitalization, respiratory support, and tube feeding. When treating pregnant women with vilazodone, consideration should be given to whether the potential benefits outweigh the potential risks of treatment (Pregnancy Category C).
The dosage of vilazodone should be reduced when the drug is coadministered with CYP3A4 strong inhibitors. Concomitant use of vilazodone with CYP3A4 inducers can result in inadequate drug concentrations and may diminish effectiveness. Vilazodone should not be used concurrently with an MAOI or within 14 days of stopping or starting an MAOI. SSRIs can increase the occurrence of upper GI bleeding, and concurrent use of a nonsteroidal anti-inflammatory drug (NSAID) or aspirin may potentiate the risk of bleeding. Altered anticoagulant effects, including increased bleeding, have been reported when SSRIs and SNRIs are coadministered with warfarin. Thus, patients taking warfarin should be carefully monitored when vilazodone is initiated or discontinued. The risk of vilazodone administered in combination with other CNS-active drugs has not been systematically evaluated; therefore, caution is advised in the case of concurrent use.
Dosage and Administration11,12: Vilazodone is available as 10-mg, 20-mg, and 40-mg tablets. The recommended dosage of vilazodone is 40 mg once daily. Treatment should be titrated, with an initial dosage of 10 mg once daily for 7 days followed by 20 mg once daily for an additional 7 days and then an increase to 40 mg once daily. No dosage adjustment is recommended in patients with mild, moderate, or severe renal impairment or with mild or moderate hepatic impairment. The vilazodone dosage should be reduced to 20 mg when the drug is coadministered with CYP3A4 strong inhibitors.
The efficacy of vilazodone has not been investigated beyond 8 weeks. Thus, patients should be reassessed periodically to determine the need for maintenance treatment and the appropriate treatment dosage.
REFERENCES
1. Edarbi (azilsartan medoxomil) product information. Deerfield, IL: Takeda Pharmaceuticals America, Inc; April 2011.
2. White WB, Weber MA, Sica D, et al. The new angiotensin receptor blocker azilsartan medoxomil has superior 24-hour blood pressure lowering efficacy to both olmesartan and valsartan [PO-242]. J Clin Hypertens. 2010;12(suppl 1):A116.
3. Pradaxa (dabigatran etexilate mesylate) product information. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals, Inc; August 2011.
4. Wallentin L, Yusuf S, Ezekowitz MD, et al. Efficacy and safety of dabigatran compared with warfarin at different levels of international normalised ratio control for stroke prevention in atrial fibrillation: an analysis of the RE-LY trial. Lancet. 2010;376:975-983.
5. Latuda (lurasidone HCl) product information. Marlborough, MA: Sunovion Pharmaceuticals Inc; October 2010.
6. Nakamura M, Ogasa M, Guarino J, et al. Lurasidone in the treatment of acute schizophrenia: a double-blind, placebo-controlled trial. J Clin Psychiatry. 2009;70:829-836.
7. Daliresp (roflumilast) product information. St. Louis, MO: Forest Pharmaceuticals, Inc; February 2011.
8. Bateman E, Holmes M, Muir JF, et al. Safety profile of roflumilast, a novel selective phosphodiesterase 4 inhibitor, in patients with moderate to severe COPD. Am J Respir Crit Care Med. 2004;169:A596.
9. Natroba (spinosad) product information. Carmel, IN: ParaPRO LLC; January 2011.
10. Stough D, Shellabarger S, Quiring J, Gabrielsen AA Jr. Efficacy and safety of spinosad and permethrin creme rinses for pediculosis capitis (head lice). Pediatrics. 2009;124:e389-e395.
11. Viibryd (vilazodone HCl) product information. St. Louis, MO: Forest Pharmaceuticals; March 2011.
12. Rickels K, Athanasiou M, Robinson DS, et al. Evidence for efficacy and tolerability of vilazodone in the treatment of major depressive disorder: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2009;70:326-333.
To comment on this article, contact rdavidson@uspharmacist.com.



