US Pharm. 2021;46(10):28-40.
New molecular entities (NMEs) are defined by the FDA as new drug products containing, as their active ingredient, a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in 2021 detail the basic clinical and pharmacologic profile for each new drug, as well as key precautions and warnings (TABLE 1). Also included is a brief summary of selected pharmacokinetic, adverse reaction, drug interaction, and dosing data submitted to the FDA in support of the manufacturer’s new drug application. This review is intended to be objective rather than evaluative in content. The information for each NME was obtained primarily from sources published prior to FDA approval. Experience clearly demonstrates that many aspects of a new drug’s therapeutic profile, not detected in premarketing studies, emerge after the drug is used in large numbers of patients. Studies have demonstrated the appearance of “new” adverse reactions for many NMEs within several years of their first becoming available. Some of these drugs may eventually acquire at least one black box warning for serious adverse drug reactions or be withdrawn from the market for safety reasons that were not recognized at the time of approval. Hence, while this review offers a starting point for learning about new drugs, it is essential that practitioners become aware of changes in a drug’s therapeutic profile as reported in the pharmaceutical literature and by their own patients.
Drospirenone and Estetrol (Nextstellis, Mayne Pharma)
Indication and Clinical Profile1,2: Drospirenone and estetrol is a new combined oral contraceptive product including the novel estrogen estetrol with drospirenone (DRSP) indicated for use by females of reproductive potential to prevent pregnancy. This is the first new estrogen introduced in the United States in more than 50 years. The efficacy of drospirenone and estetrol was evaluated in a multicenter, open-label, single-arm study in North America of 1-year duration that enrolled 1,674 females aged 16 to 35 years. The mean age was 25.8 years, and mean BMI was 25.8 kg/m2. Females with a BMI between 30 and 35 kg/m2 accounted for 22.3% of the study population. The racial distribution was 70.1% Caucasian, 19.5% Black or African American, 4.8% Asian, 0.9% American Indian or Alaska native, 0.4% Native Hawaiian or other Pacific Islander, and 4.2% other. A total of 26 on-treatment pregnancies occurred in 1,524 females contributing 12,763 at-risk cycles. The overall Pearl Index (statistical estimation of the number of unintended pregnancies in 100 woman-years of exposure) was 2.65 per 100 woman-years of use. A trend of decreasing effectiveness with increasing BMI was observed in the study. Clinical results showed a desirable bleeding profile and minimal impact on triglycerides, cholesterol, and glucose and on weight and endocrine markers. Drospirenone and estetrol also produced significantly lower increases in sex hormone-binding globulin compared with two other contraceptives containing ethinyl-estradiol.
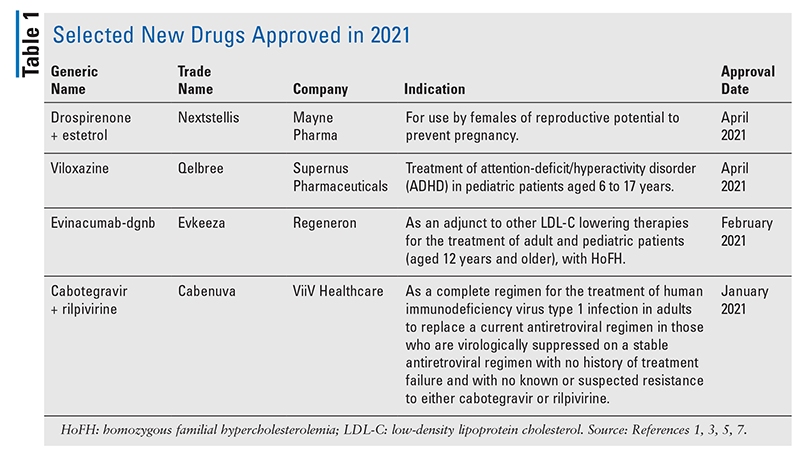
Pharmacology and Pharmacokinetics1,2: Drospirenone is a spironolactone analogue with antimineralocorticoid and antiandrogenic activity. The estrogen component is estetrol, a synthetic trihydroxy analogue of a native estrogen (FIGURE 1) present during pregnancy, which is selective for nuclear estrogen receptor (ER)-alpha and ER-beta. Like other contraceptives, this drug product prevents pregnancy primarily by suppressing ovulation.
Both components of the drospirenone and estetrol product provide peak plasma levels within an hour after oral administration. Food does not significantly alter plasma concentrations. Estetrol is only 46% to 50% bound by plasma proteins, while drospirenone is 96% bound. Estetrol undergoes conjugative metabolism to form glucuronide and sulphate conjugates. Drospirenone is metabolized by CYP3A4 to yield an acid metabolite by opening of the lactone ring, and 4,5-dihydrodrospirenone is formed by reduction followed by sulfation. Estetrol is primarily excreted in the urine while drospirenone is excreted in both the urine and feces.
Adverse Reactions and Drug Interactions1,2: The most common adverse reactions (≥2%) linked to drospirenone and estetrol include bleeding irregularities, mood disturbance, headache, breast symptoms, dysmenorrhea, acne, weight gain, and decreased libido. The drug should be discontinued if blood pressure rises significantly or is persistent, or if severe migraines occur. Drospirenone and estetrol should also be discontinued if thromboembolic events occur or if hormonally sensitive malignancy or symptomatic gallbladder or cholestatic disease is diagnosed. Due to the potential to cause hyperkalemia, serum potassium concentrations should be determined during the first treatment cycle in patients on long-term treatment with medications that may increase serum potassium concentration.
The drospirenone and estetrol product contains a warning stating that cigarette smoking increases the risk of serious cardiovascular events from combined hormonal contraceptive use. This risk increases with age, particularly in females over age 35 years, and with the number of cigarettes smoked. Combined hormonal contraceptives, including drospirenone and estetrol, are contraindicated in females who are older than age 35 years and smoke. The drug is also contraindicated in patients with a high risk of arterial or venous thrombotic diseases, current or history of a hormonally sensitive malignancy (e.g., breast cancer), hepatic adenoma, hepatocellular carcinoma, acute hepatitis or decompensated cirrhosis, abnormal uterine bleeding that has an undiagnosed etiology, renal impairment, or adrenal insufficiency. Also, drospirenone and estetrol should not be coadministered with hepatitis C drug combinations containing ombitasvir/paritaprevir/ritonavir, with or without dasabuvir.
Estetrol is not a substrate for any of the major CYP isozymes or drug transporters at clinically relevant doses. Coadministration of drospirenone and estetrol with CYP3A inducers may result in enhanced drug inactivation leading to contraceptive failure and/or increase breakthrough bleeding. If concomitant use of drospirenone and estetrol with CYP3A inducers is unavoidable, an alternative or backup contraceptive method should be used during coadministration and up to 28 days after discontinuation. Also concomitant use with bile-acid sequestrants may decrease the exposure of both drugs in this product, which may lead to contraceptive failure and/or an increase in breakthrough bleeding. Drospirenone is a CYP3A4 substrate and therefore concomitant use with strong CYP3A inhibitors may increase drospirenone exposure, which may increase the risk of adverse reactions including hyperglycemia.
Dosage and Administration1,2: Drospirenone and estetrol is available in a blister card, with 28 film-coated tablets including 24 pink active tablets each containing drospirenone 3 mg and estetrol 14.2 mg, and four white inert tablets. It should be initiated using a Day 1 start, and one tablet by mouth should be taken in the order directed on the blister pack at the same time every day. The drug may be administered with or without food. Drospirenone and estetrol is contraindicated in females with renal or hepatic impairment.
Viloxazine Extended-Release Capsules (Qelbree, Supernus Pharmaceuticals)
Indication and Clinical Profile3,4: Viloxazine extended-release capsules are indicated for the treatment of attention-deficit/hyperactivity disorder (ADHD) in pediatric patients aged 6 to 17 years. The FDA approval of viloxazine extended-release capsules was based on three short-term, randomized, placebo-controlled monotherapy trials (studies 1, 2, and 3). Study 1 was a multicenter, randomized, double-blind, three-arm, placebo-controlled, parallel-group monotherapy trial in patients aged 6 to 11 years with ADHD. A total of 477 patients were randomized; 399 completed the study, and 78 discontinued. The duration of the trial was 6 weeks, including a 1-week titration period (starting at 100 mg once daily) and 5-week maintenance phase. Patients were randomized to receive 100 mg, 200 mg, or placebo, given once daily as a single dose. The primary endpoint was the change from baseline to the end of study on the total score on the ADHD Rating Scale (ADHD-RS-5), an 18-question scale that assesses hyperactivity, impulsivity, and inattention symptoms, where higher scores reflect more severe symptoms. A key secondary endpoint was the Clinical Global Impression–Improvement (CGI-I) score at the end of the study. The change from baseline (reduction) in ADHD-RS-5 total score was statistically significantly greater in patients treated with 100 or 200 mg viloxazine extended-release capsules than in patients on placebo. Compared with patients on placebo, a statistically significantly greater reduction (improvement) in CGI-I score at the end of the study was observed in patients treated with both doses of viloxazine extended-release capsules. Similar results using the same endpoints were obtained in Study 2 and Study 3, both multicenter, randomized, double-blind, placebo-controlled monotherapy trials in patients aged 6 to 11 years (Study 2) or aged 12 to 17 years (Study 3) with ADHD. Each of these trials involved over 300 patients treated for 6 weeks (Study 3) or 8 weeks (Study 2).
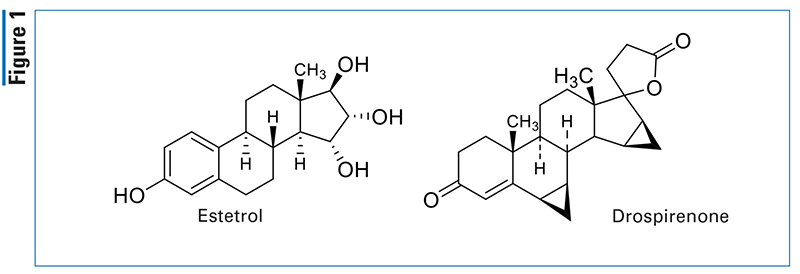
Pharmacology and Pharmacokinetics3,4: Viloxazine extended-release capsules is a racemic-2 [(2-ethoxyphenoxy)methyl]-morpholine derivative (FIGURE 2) that acts as a selective norepinephrine reuptake inhibitor. The mechanism of action in the treatment of ADHD is unclear; however, it is thought to occur through inhibition of the norepinephrine reuptake transporter.
The relative bioavailability of viloxazine extended-release relative to an immediate-release formulation is about 88%, giving peak plasma levels in about 5 hours. Viloxazine is primarily metabolized by CYP2D6, UGT1A9, and UGT2B15. The major metabolite detected in plasma is 5-hydroxy-viloxazine glucuronide. Renal excretion is the primary route of excretion (90%) of viloxazine. The mean half-life of viloxazine is reported to be about 7 hours.
Adverse Reactions and Drug Interactions3,4: The most common adverse reactions (≥5% and at least twice the rate of placebo) associated with use of viloxazine extended-release capsules include somnolence, decreased appetite, fatigue, nausea, vomiting, insomnia, and irritability. The drug can also cause an increase in heart rate and diastolic blood pressure, and these parameters should be assessed prior to initiating treatment. The drug label carries a black boxed warning for suicidal thoughts and behaviors. In clinical trials, higher rates of suicidal thoughts and behavior were reported in pediatric patients treated with viloxazine extended-release capsules than in patients treated with placebo. Based on these observations, close monitoring for worsening and emergence of suicidal thoughts and behaviors is recommended.
Viloxazine extended-release capsules may induce a manic or mixed episode in patients with bipolar disorder, similar to other noradrenergic medications. Prior to initiating treatment, patients should be screened for risk, screening should include a detailed psychiatric history, including a personal or family history of suicide, bipolar disorder, and depression. Also, viloxazine extended-release capsules can cause somnolence and fatigue, and patients should be counseled to not perform activities requiring mental alertness, such as operating a motor vehicle or operating hazardous machinery, until they are aware of how they will be affected by this drug.
Viloxazine extended-release capsules is contraindicated in patients receiving concomitant treatment with monoamine oxidase inhibitors (MAOI), or within 14 days following discontinuing an MAOI, due to an increased risk of hypertensive crisis. It is also contraindicated in patients receiving concomitant administration of sensitive CYP1A2 substrates or CYP1A2 substrates with a narrow therapeutic range, since viloxazine extended-release capsules is a potent CYP1A2 inhibitor.
Based on findings from animal reproduction studies, viloxazine may cause maternal harm when used during pregnancy. Discontinue viloxazine extended-release capsules when pregnancy is recognized unless the benefits of therapy outweigh the potential risk to the mother. There are no data on the presence of viloxazine in human milk, the effects on the breastfed infant, or the effects on milk production.
Dosage and Administration3,4: Viloxazine extended-release capsules is supplied as 100-, 150-, and 200- mg extended-release capsules for oral administration. They can be administered with or without food. Capsules should be swallowed whole or opened and sprinkled over a teaspoonful of applesauce and swallowed. Capsules should not be cut, crushed or chewed. The recommended dose for pediatric patients aged 6 to 11 years is 100 mg orally once daily. Dosage may be titrated in increments of 100 mg at weekly intervals to the maximum recommended dosage of 400 mg once daily, depending on response and tolerability. The recommended starting dosage for pediatric patients aged 12 to 17 years is 200 mg orally once daily. After 1 week, dosage may be titrated by an increment of 200 mg to the maximum recommended dosage of 400 mg once daily, depending on response and tolerability. No dosage adjustment is recommended in patients with mild to moderate renal impairment (eGFR of 30-89 mL/min/1.73m2). However, for patients with severe renal impairment (eGFR <30 mL/min/1.73m2), the starting dosage is 100 mg once daily. The pharmacokinetics of viloxazine have not been evaluated in hepatic impairment. Pharmacologic treatment of ADHD may be required for extended periods, and the long-term use should be reevaluated periodically and dosage adjusted as needed.
Evinacumab-dgnb (Evkeeza, Regeneron)
Indication and Clinical Profile5,6: Evinacumab-dgnb is specifically indicated as an adjunct to other low-density lipoprotein cholesterol (LDL-C)-lowering therapies for the treatment of adult and pediatric patients (aged 12 years and older) with homozygous familial hypercholesterolemia (HoFH). The FDA approval of evinacumab-dgnb was based on results from ELIPSE HoFH, a phase III randomized, double-blind, placebo-controlled trial evaluating the efficacy and safety of evinacumab-dgnb 15 mg/kg administered IV every 4 weeks in 65 HoFH patients aged 12 years or older. The primary endpoint was reduction of LDL-C after 24 weeks. Results from the evinacumab-dgnb group at week 24 included a 49% reduction in LDL-C from baseline, compared with placebo. Evinacumab-dgnb–treated patients also had a 132–mg/dL absolute change in LDL-C from baseline, compared with placebo, and 47% of patients in the evinacumab-dgnb arm achieved LDL- C levels less than 100 mg/dL, compared with 23% for placebo. Similar levels of LDL-C lowering were also observed in the most difficult-to-treat patients (“null/null” or “negative/negative”patients) who often do not respond to certain other therapies. Evinacumab-dgnb also reduced apolipoprotein B, non–high-density lipoprotein cholesterol (HDL-C), and total cholesterol compared with placebo. The safety and effectiveness of evinacumab-dgnb have not been established in patients with other causes of hypercholesterolemia, including those with heterozygous familial hypercholesterolemia, and the effects of evinacumab-dgnb on cardiovascular morbidity and mortality have not been determined.
Pharmacology and Pharmacokinetics5,6: Evinacumab-dgnb is a recombinant human monoclonal antibody that binds to and inhibits angiopoietin-like 3 (ANGPTL3), a member of the angiopoietin-like protein family that is expressed primarily in the liver and plays a role in the regulation of lipid metabolism by inhibiting lipoprotein lipase (LPL) and endothelial lipase (EL). Evinacumab-dgnb inhibition of ANGPTL3 results in a reduction in LDL-C, HDL-C, and triglycerides (TG). Evinacumab-dgnb reduces LDL-C independent of the presence of LDL receptor by promoting very low-density lipoprotein processing and clearance upstream of LDL formation. Evinacumab-dgnb inhibition of ANGPTL3 also lowers TG and HDL-C by rescuing LPL and EL activities, respectively.
Steady-state plasma levels of evinacumab-dgnb are reached after four doses, and the total volume of distribution is 4.8 L. The drug is eliminated by parallel linear and nonlinear pathways. At higher concentrations, evinacumab-dgnb elimination is primarily through a nonsaturable proteolytic pathway, whereas at lower concentrations, the nonlinear, saturable ANGPTL3 target-mediated elimination predominates. The elimination half-life is a function of serum evinacumab-dgnb concentrations and is not a constant. The median time for drug concentrations to decrease below the lower limit of quantitation is 19 weeks after the last steady-state dose of 15 mg/kg IV every 4 weeks. Although the pathway of metabolism has not been determined, evinacumab-dgnb is expected to be degraded into small peptides and amino acids via catabolic pathways in the same manner as endogenous immunogloblin G (IgG). Serum concentrations at steady state are comparable in patients with mild-to-moderate renal impairment and patients with normal renal function, thus dose adjustments based on renal function are not anticipated. At present no data are available in patients with severe renal impairment or in patients with hepatic impairment.
Adverse Reactions and Drug Interactions5,6: In clinical trials, the primary adverse effects associated with the use of evinacumab-dgnb included nasopharyngitis, influenza-like illness, dizziness, rhinorrhea, and nausea. Serious hypersensitivity reactions have been reported in clinical trials. If serious hypersensitivity reactions occur, evinacumab-dgnb should be discontinued and patients treated and monitored according to standard of care. Evinacumab-dgnb may cause fetal harm based on data from animal studies. Thus, pregnancy testing is recommended prior to initiating treatment, and patients who may become pregnant should be advised to use contraception during treatment and for at least 5 months following the last dose. There are no data on the presence of evinacumab-dgnb in human milk or on the effects on the breastfed infant, but maternal IgG is known to be present in human milk.
Formal drug interaction studies have not been conducted with evinacumab-dgnb. In a clinical trial, the concentrations of statins (atorvastatin, rosuva-statin, simvastatin) were not significantly altered in patients taking statins prior to and post administration of evinacumab-dgnb.
Dosage and Administration5,6: Evinacumab-dgnb is supplied as a 345 mg/2.3 mL (150 mg/mL) and 1,200 mg/8 mL (150 mg/mL) solution for IV administration. Other medications should not be mixed with evinacumab-dgnb or administered concomitantly via the same infusion line. The recommended dose is 15 mg/kg administered by IV infusion over 60 minutes every 4 weeks. If a dose is missed, it should be administered as soon as possible. Thereafter, doses should be scheduled monthly from the date of the last dose. The LDL-C–lowering effect of evinacumab-dgnb may be measured as early as 2 weeks after initiation.
Cabotegravir and Rilpivirine (Cabenuva, ViiV Healthcare)
Indication and Clinical Profile7,8: Cabotegravir and rilpivirine injectible formulation is a once-monthly, extended-release regimen combining the new integrase strand transfer inhibitor cabotegravir with rilpivirine, a nonnucleoside reverse transcriptase inhibitor. This product is specifically indicated as a complete regimen for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in adults to replace a current antiretroviral regimen in those who are virologically suppressed on a stable antiretroviral regimen with no history of treatment failure and with no known or suspected resistance to either cabotegravir or rilpivirine.
The FDA approval of cabotegravir and rilpivirine was based on the pivotal phase III ATLAS (Antiretroviral Therapy as Long-Acting Suppression) and FLAIR (First Long-Acting Injectable Regimen) trials that included more than 1,000 patients from 16 countries, including the United States. In these studies, cabotegravir and rilpivirine was found to be as effective as continuing a daily, oral, three-drug regimen in maintaining viral suppression throughout the 48-week study period. In the ATLAS study, cabotegravir and rilpivirine met the primary endpoint for noninferiority, with a comparable number of patients receiving either cabotegravir and rilpivirine or their daily current antiretroviral regimen (CAR) having an HIV-1 RNA level greater than or equal to 50 c/mL; only 2% of patients receiving cabotegravir and rilpivirine and 1% of patients receiving CAR had an HIV-1 RNA level greater than or equal to 50 c/mL at Week 48. In the FLAIR study, at Week 48, a comparable number of patients receiving either cabotegravir and rilpivirine or daily oral therapy with the combination of dolutegravir/abacavir/lamivudine had an HIV-1 RNA count greater than or equal to 50 c/mL, meeting noninferiority criteria. Only 2% of patients in both treatment arms had an HIV-1 RNA count greater than or equal to 50 c/mL at Week 48.
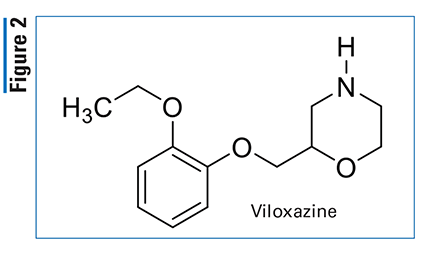
Pharmacology and Pharmacokinetics7,8: The cabotegravir and rilpivirine product contains the novel oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide integrase strand transfer inhibitor (INSTI) cabotegravir with the nonnucleoside reverse transcriptase inhibitor rilpivirine (FIGURE 3). INSTIs inhibit HIV replication by preventing the viral DNA from integrating into the genome of human T-cells. This step is essential in the HIV replication cycle and is also responsible for establishing chronic infection. Rilpivirine inhibits the HIV enzyme reverse transcriptase, which in turn prevents the virus from multiplying.
Both cabotegravir and rilpivirine are well absorbed from injection sites, achieving maximum plasma levels at 7 days and 3 to 4 days, respectively. The drugs are well distributed and highly bound by plasma proteins (>99%). Cabotegravir is cleared by UGT1A1 conjugation while rilpivirine is metabolized by CYP3A4. Both drugs are excreted primarily by the liver with elimination half-lives of 5.6 to 11.5 hours for cabotegravir and 13 to 28 hours for rilpivirine.
Adverse Reactions and Drug Interactions7,8: The most common adverse reactions (Grades 1 to 4) observed in 2% or more of subjects receiving cabotegravir and rilpivirine were injection-site reactions, pyrexia, fatigue, headache, musculoskeletal pain, nausea, sleep disorders, dizziness, and rash. Hypersensitivity reactions have been reported with rilpivirine-containing regimens and in association with other integrase inhibitors. Cabotegravir and rilpivirine therapy should be discontinued immediately if signs or symptoms of hypersensitivity reactions develop. Hepatotoxicity has been reported in patients receiving cabotegravir or rilpivirine; thus, monitoring of liver function is recommended, and discontinuation if hepatotoxicity is suspected. Depressive disorders (including depressed mood, depression, major depression, altered mood, dysphoria, negative thoughts, attempted suicide, or suicidal ideation) have been associated with cabotegravir and rilpivirine and the individual drug products.
Cabotegravir and rilpivirine are detected in systemic circulation for up to 12 months or longer after discontinuing injections of cabotegravir and rilpivirine; therefore, consideration should be given to the potential for fetal exposure during pregnancy. There are insufficient human data on the use of cabotegravir and rilpivirine during pregnancy to adequately assess a drug-associated risk of birth defects and miscarriage. Neural tube defects were associated with another integrase inhibitor (dolutegravir); thus, the benefit-risk of using cabotegravir and rilpivirine should be discussed with individuals of childbearing potential or during pregnancy.
Because cabotegravir and rilpivirine is a complete HIV regimen, coadministration with other antiretroviral medications is not recommended. Drugs that induce uridine diphosphate glucuronosyl-transferase (UGT)1A1 or cytochrome 3A4, including carbamazepine, oxcarbazepine, phenobarbital, phenytoin, the rifampins, glucocorticoids, and St. John’s wort, may decrease the plasma concentrations of the components of cabotegravir and rilpivirine resulting in loss of virologic response. Cabotegravir and rilpivirine should be used with caution in combination with drugs with a known risk of torsade de pointes, such as macrolides.
Dosage and Administration7,8: Cabotegravir and rilpivirine should be administered by a healthcare provider once monthly as two individual IM gluteal injections. Prior to initiating treatment with cabotegravir and rilpivirine, oral cabotegravir (Vocabria) and oral rilpivirine (Edurant) should be administered for approximately 1 month to assess the tolerability of each therapy. Injections of 600 mg of cabotegravir and 900 mg of rilpivirine should be started on the last day of oral lead-in and continue with injections of 400 mg of cabotegravir and 600 mg of rilpivirine every month thereafter.
Based on studies with oral cabotegravir and population pharmacokinetic analyses of oral rilpivirine, no dosage adjustment of cabotegravir and rilpivirine is necessary for patients with mild or moderate renal impairment or mild or moderate hepatic impairment. In patients with severe renal impairment (creatinine clearance 15-30 mL/min) or end-stage renal disease (creatinine clearance <15 mL/min), increased monitoring for adverse effects is recommended. The effect of severe hepatic impairment (Child-Pugh C) on the pharmacokinetics of cabotegravir or rilpivirine is not known.
REFERENCES
1. Nextstellis (drospirenone and estetrol tablets) package insert. Greenville, NC: Mayne Pharma. April 2021.
2. Bennink C Holinka CF, Visser M, et al. Maternal and fetal estetrol levels during pregnancy. Climacteric. 2008. 11(Suppl 1):69-72.
3. Qelbree (viloxazine extended-release capsules) package insert. Rockville, MD: Supernus Pharmaceuticals Inc. April 2021.
4. Supernus Pharmaceuticals, Inc. Supernus announces FDA approval of Qelbree (SPN-812) for the treatment of ADHD. April 2, 2021. https://ir.supernus.com/news-releases/news-release-details/supernus-announces-fda-approval-qelbreetm-spn-812-treatment-adhd. Accessed September 10, 2021.
5. Evkeeza (evinacumab-dgnb) injection package insert. Tarrytown, NY: Regeneron Pharmaceuticals, Inc. February 2021.
6. Raal FJ, Rosenson RS, Reeskamp L F, et al. Evinacumab for homozygous familial hypercholesterolemia, N Engl J Med. 2020;383:711-720.
7. Cabenuva (cabotegravir extended-release injectable suspension; rilpivirine extended-release injectable suspension, copackaged for intramuscular use) package insert. Research Triangle Park, NC: ViiV Healthcare; January 2021.
8. Swindells S, Andrade-Villanueva JF A, Richmond, GJ, et al. Long-acting cabotegravir and rilpivirine for maintenance of HIV-1 Suppression. N Engl J Med. 2020;382:1112-1123.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
To comment on this article, contact rdavidson@uspharmacist.com.


