US Pharm. 2015;40(7)(Specialty&Oncology suppl):4-7.
ABSTRACT: Sickle cell disease (SCD), a hereditary blood disorder that causes the body to produce abnormal sickle hemoglobin (HbS), has become the driving force for a series of resultant medical complications. Such clinical manifestations include anemia via hemolysis, iron overload secondary to repeated blood transfusions, bacterial infections, pulmonary complications, vascular occlusion, and episodic pain crises. Preventative therapy most commonly consists of hydroxyurea, routine vaccinations, and prophylactic antibiotics. However, iron chelation therapy and pain management are considered mainstay treatment options as the disease persists. Because SCD can drastically reduce quality of life and life expectancy, it is vital that the pharmacist plays a fundamental role in providing an optimal treatment and management plan for each individual patient combating this disease.
Sicke cell disease (SCD) is caused by a single amino acid substitution at the sixth position from glutamic acid to valine on the beta-globin chain of adult hemoglobin (HbA).1 Such substitution leads to polymerization of oxygen-poor sickle hemoglobin (HbS), thus producing sickle-shaped red blood cells. This results in a sequelae of complications including acute chest syndrome (ACS), anemia, chronic pain, pneumococcal infections, pulmonary hypertension, ischemic complications, and vaso-occlusive crises (VOCs).1
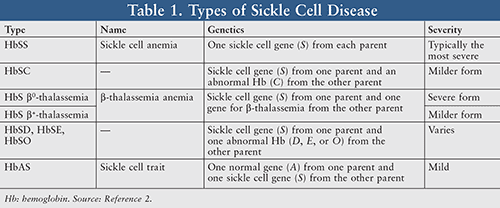
While SCD was first medically described in 1910, it was further explained on a more precise genetic and molecular level decades later. This hereditary blood disorder has been classified into an array of types or subtypes contingent upon an individual’s gene inheritance from each parent (TABLE 1).2
Although SCD affects millions of people worldwide, it is predominantly prevalent among persons of Sub-Saharan African, South American, Caribbean, Central American, Saudi Arabian, Indian, and Mediterranean ancestry.2 According to the CDC, an estimated 90,000 to 100,000 Americans, mainly of African-American heritage, are affected by SCD. Roughly one in 12 African Americans will inherit the sickle cell trait from one parent.
In 2013, it was reported that complications from this disease resulted in 176,000 deaths, compared to 113,000 deaths in 1990.3 Patients with SCD often present with numerous complications, both acute and chronic, which may lead to hospitalizations, morbidity, and, ultimately, death. Of the many complications associated with SCD, this article will focus on the review and management of anemia, bacterial predispositions, pulmonary complications, VOCs, and pain.
Clinical Manifestations
Anemia: The transfusion of normal red blood cell donations containing HbA into patients with SCD significantly reduces the percentage of circulating HbS-containing red blood cells. Such transfusions are utilized to mitigate the unfavorable effects of sickled erythrocytes. However, the risk of erythrocyte alloimmunization and hyperhemolysis increases with the frequency of blood transfusions, resulting in serious consequences for the patient. At times, it can be difficult to locate compatible units, thereby resulting in transfusion delays.4
In SCD patients, iron overload is attributable to repeated blood transfusions. Treatment and monitoring of transfusional iron overload in SCD is essential to preventing myocardial and hepatic iron disposition, as well as to reducing the risk of cirrhosis and hepatocellular carcinoma. Serum ferritin levels and MRI techniques are used to monitor the liver iron concentration (LIC) levels, minimize over- or underchelation, and measure the response to chelation.5
SCD Bacterial Predispositions: Those with SCD have a greater predisposition to severe bacterial infections as a result of their reduced or absent splenic function due to entrapment of sickled erythrocytes in the spleen.1 Streptococcus pneumoniae and Haemophilus influenzae commonly infect children because of their unusual susceptibility to these pathogens caused by splenic malfunction and failure to make specific immunoglobulin G (IgG) antibodies to polysaccharide antigens.6 Young children diagnosed with SCD have a higher risk of contracting an invasive pneumococcal disease caused by S pneumoniae, often resulting in increased septicemia and meningitis. Adults, in contrast, present without fever (afebrile), shortness of breath, chills, and severe pain. Pathogens including Staphylococcus aureus and Salmonella, Enterobacter, and Clostridium species are detected among older individuals.6
Pulmonary Complications: Borderline/mild pulmonary hypertension (pulmonary artery systolic pressure >35 mm) is associated with a high risk of death in patients diagnosed with SCD. Brain natriuretic peptide (BNP), which is released from cardiomyocytes during pressure or volume stretch, is also an indicator of pulmonary hypertension. Iron overload, hepatitis C, nodular hepatic regenerative hyperplasia resulting in liver dysfunction, and chronic renal failure have been identified as mechanisms contributing to the development of pulmonary hypertension in patients with SCD.7 It is suggested that acute pulmonary hypertension and right heart dysfunction are major coexisting conditions in the acute syndrome.7 Identification, prevention, and management of these pulmonary complications can lead to increased life expectancy.
ACS is defined by the development of a new pulmonary infiltrate that is consistent with alveolar consolidation involving at least one complete lung segment. It is considered to be the second most common cause of hospitalization and the leading cause of ICU admission and premature death (up to 25%) in the SCD patient population.6,7 ACS typically develops 24 to 72 hours after the onset of severe pain in the arms, legs, and chest of most adult patients with sickle cell anemia.7 The major proposed causes of ACS are pulmonary infection by a community-acquired pathogen, embolization of bone marrow fat (associated with painful VOCs involving multiple bones like the pelvis and femur), and intravascular pulmonary sequestration of sickled erythrocytes.4 As the presenting symptoms are age-dependent, the clinical presentation and course differ between children and adults. The death rate is reported to be four times higher in adults than in children due to lower or multilobe involvement, severe hypoxia, pulmonary thrombotic events, and longer hospitalizations.6
Vascular Occlusion: Complications associated with vascular occlusion have reportedly been the cause of the decreased lifespan in those affected by SCD.1 VOCs, or acute pain crises, are defined by unpredictable recurrent episodes of severe bouts of pain in a person with SCD and require carefully controlled pain management. This complication is derived from the microvascular entrapment of erythrocytes and leukocytes, thereby causing an obstruction of blood flow resulting in organ ischemia.7 VOC serves as a risk factor for consequential complications, most notably ACS.1
Preventative Therapy
Hydroxyurea: Hydroxyurea has earned its title as the mainstay treatment for those with SCD. This oral ribonucleotide reductase inhibitor has been proven to induce fetal hemoglobin levels and inhibit the polymerization of HbS, thus reducing and preventing acute and chronic SCD-associated complications.1 Additionally, the nitric oxide released from the metabolism of hydroxyurea may be an advantageous contributor to vasodilation, which is needed to prevent a hypercoagulable state.1 The long-term use of hydroxyurea has demonstrated the ability to reduce the amount of pain crises, ACS events, red blood cell transfusion requirements, hospitalizations, and death.1,8
Hydroxyurea therapy should be offered in both asymptomatic and symptomatic children ≥9 months of age.1 When utilized in SCD, hydroxyurea is generally initiated at an oral dose of 15 mg/kg and titrated by 5 mg/kg/day every 12 weeks with a maximum dose of 35 mg/kg/day (TABLE 2).8 Common side effects of this medication include myelosuppression, erythrocyte abnormalities, elevated liver enzymes, renal impairment, and a high potential for fetal risk. Therefore, drug-monitoring parameters should consist of a CBC with differential (target ANC ≥2,000/mcL), platelet counts (maintain ≥80,000/mcL), a quantitative measurement of fetal hemoglobin, renal and liver function tests, and a pregnancy test for women.8
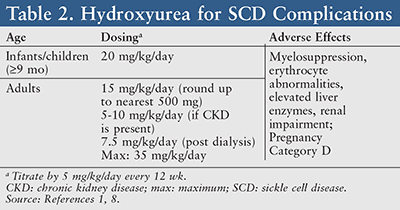
Hydroxyurea (Pregnancy Category D) is generally not prescribed for pregnant or lactating women, as this drug is known to affect DNA synthesis via its mechanism of action and has the ability to cross the placenta.8 However, recent findings suggest that human pregnancies have not experienced such teratogenic changes resulting from exposure of the fetus to hydroxyurea.9 A clinical response may take up to 3 to 6 months to be seen; therefore, it is imperative to encourage patient adherence.
Vaccines and Prophylactic Antibiotics: Pneumococcal vaccines and prophylactic antibiotics (TABLE 3) are used as preventative strategies for S pneumoniae, which is the most common cause of death in children with SCD.6 It is also recommended that adults receive preventative pneumococcal vaccines, in addition to an annual influenza vaccine.6 For pneumococcal infection prophylaxis, oral penicillin VK twice daily is the antibiotic of choice for children. As a supplement to the antibiotics, scheduled pneumococcal vaccines should be given to children according to age. Prevnar 13 (PCV 13) is administered as a one-time 0.5 mL intramuscular (IM) injection to those aged >6 years, whereas Pneumovax 23 (PPSV 23) is given to those aged ≥19 years as a one-time 0.5 mL subcutaneous (SC) or IM injection along with PCV 13 administered >8 weeks beforehand. This immunization schedule must be repeated once again in 5 years. Influenza vaccines are typically given as an 0.5 mL IM injection once a year, and product selection may vary according to preference or availability.1

Treatment Options
Iron Chelation Therapy: Removal of excess iron reduces vital organ and tissue damage, in addition to increasing the recovery time of nerve damage or trauma.5 There are currently three medications available for iron chelation: deferoxamine, deferasirox, and deferiprone (TABLE 4). Among the available options, deferoxamine is the only iron chelator parenterally administered as a daily dose of 0.5 to 1 g IM or by slow SC or IV infusion.5 The relatively long half-life (8-16 hours) of deferasirox, the first oral iron chelation medication approved in the United States, allows for convenient once-daily dosing of this tablet. In comparison, deferiprone is an oral film-coated tablet with a shorter half-life (2-3 hours) that can be more costly due to the required effective frequency of administration (three times a day).5
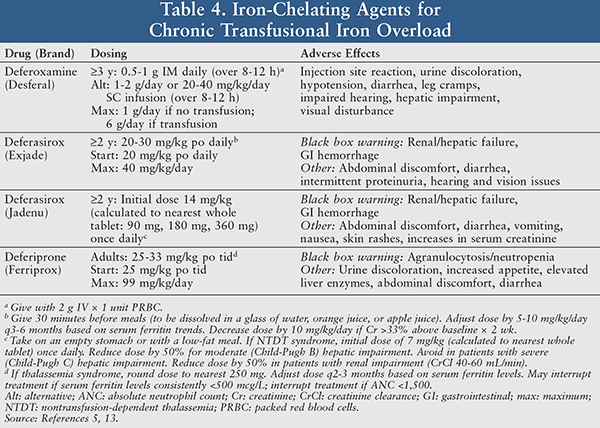
Notable side effects of this drug class include urine discoloration, diarrhea, and hepatic impairment. Drug-associated black box warnings such as renal/hepatic failure and gastrointestinal (GI) hemorrhage for deferasirox and agranulocytosis/neutropenia for deferiprone require close monitoring of the patient’s laboratory values to ideally prevent or readily treat such adverse effects.5 The decision between each agent of choice for iron chelation is multi-factorial and includes route of administration, cost, and overall patient preference.
Pain Management: Pain management in SCD patients often refers to proper analgesia employed for relief of both painful acute VOCs and chronic intermittent or persistent pain. The use of analgesics should routinely be individualized and tailored to mitigate the patient’s level of pain with a primary goal of providing the patient with adequate pain relief. The therapeutic decision between nonopioid medications and opioids is dependent upon the pain severity level and risk-benefit patient assessment (TABLE 5).10
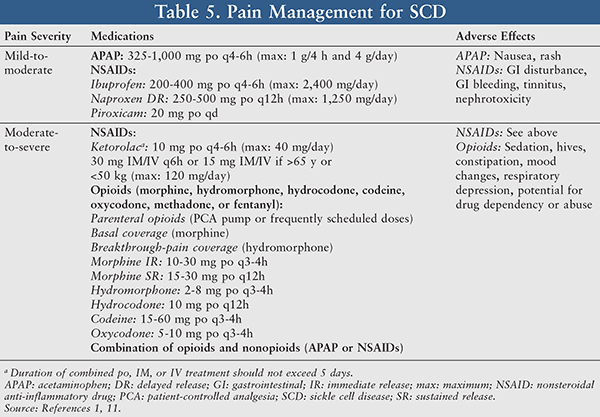
Acetaminophen and nonsteroidal anti-inflammatory drugs (NSAIDs; e.g., ibuprofen, piroxicam, and ketorolac) are typically utilized for treatment of mild-to-moderate pain. When assessing a patient’s candidacy for these drugs, it is important to note that SCD patients have varying degrees of hepatic impairment and renal impairment, thereby resulting in a contraindication with acetaminophen. In addition, if the patient has gastritis, peptic ulcers, coagulation disorders, or renal failure, he or she is not a candidate for NSAIDs.10,11 Common side effects associated with NSAID use include GI bleeding, nephrotoxicity, and tinnitus.
For moderate-to-severe pain, a patient can be treated with either opioids (morphine, hydromorphone, codeine, hydrocodone, oxycodone, methadone, or fentanyl) or a combination of opioids and nonopioids (acetaminophen or NSAIDs).11 Morphine is considered the drug of choice when treating acute sickle cell pain crises.11 It has been reported that the clearance of morphine in SCD patients was 3- to 10-fold higher than in the non-SCD population.12 The AUC and half-life were also lower in SCD patients; however, the volume of distribution did not vary. Morphine’s pharmacokinetic characteristics of accelerated clearance and poor opioid response are likely attributable to the need for higher doses and greater frequency of opioids in order to achieve plasma levels comparable to those of patients without SCD.12 Sedation, hives, constipation, mood changes (dysphoria/euphoria), and more severe adverse effects such as respiratory depression and dependency or abuse have been reported with opioid use.1
Healthcare providers are faced with the issue of distinguishing between patients receiving proper management of pain versus those who exhibit characteristics of opioid addiction or drug-seeking behavior. For opioids in particular, comprehensive knowledge of increased clearance and correspondingly lower plasma exposures in SCD is necessary in order to effectively select appropriate doses and frequency of their administration.12
Conclusion
A drastic reduction in quality of life and a decrease in life expectancy have been associated with SCD; thus, it is imperative that such patients be adequately treated and well managed.1 A multidisciplinary healthcare team approach is often utilized to care for patients with SCD. Within that team, the pharmacist has the opportunity to play an essential role by ensuring optimal medication therapy for each individual patient.
REFERENCES
1. Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014; 312(10):1033-1048.
2. CDC; National Center on Birth Defects and Developmental Disorders: Division of Blood Disorders. Sickle cell disease (SCD): facts about sickle cell disease. www.cdc.gov/ncbddd/sicklecell/facts.html. Accessed February 28, 2015.
3. GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;385(9963):117-171.
4. Yazdanbakhsh K, Ware RE, Noizat-Pirenne F. Red blood cell alloimmunization in sickle cell disease: pathophysiology, risk factors, and transfusion management. Blood. 2012;120(3):528-537.
5. Porter J, Garbowski M. Consequences and management in iron overload in sickle cell disease. Hematology Am Soc Hematol Educ Program. 2013;2013(1):447-456.
6. Vichinsky EP, Styles LA, Colangelo LH, et al. Acute chest syndrome in sickle cell disease: clinical presentation and course. Cooperative Study of Sickle Cell Disease. Blood. 1997;89(5):1787-1792.
7. Gladwin MT, Vichinsky E. Pulmonary complications of sickle cell disease. N Engl J Med. 2008;359(21):2254-2265.
8. Charache S, Terrin ML, Moore RD, et al; Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. N Engl J Med. 1995;332(20): 1317-1322.
9. Ballas SK, McCarthy WF, Guo N, et al. Exposure to hydroxyurea and pregnancy outcomes in patients with sickle cell anemia. J Natl Med Assoc. 2009;101(10):1046-1051.
10. National Institutes of Health; National Heart, Lung, and Blood Institute; Division of Blood Diseases and Resources. The Management of Sickle Cell Disease. NIH pub no. 02-2117. Revised June 2002 (4th ed). www.nhlbi.nih.gov/files/docs/guidelines/sc_mngt.pdf. Accessed April 10, 2015.
11. Terrie YC. Improving pain management for the sickle cell patient. Pharmacy Times. September 15, 2009. www.pharmacytimes.com/publications/issue/2009/September2009/rxfocussicklecell. Accessed March 20, 2015.
12. Darbari DS, Neely M, van den Anker J, Rana S. Increased clearance of morphine in sickle cell disease: implications for pain management. J Pain. 2011;12(5):531-538.
13. Jadenu (deferasirox) package insert. East Hanover, NJ: Novartis Pharmaceuticals Corporation; March 2015. www.pharma.us.novartis.com/product/pi/pdf/jadenu.pdf. Accessed June 3, 2015.
To comment on this article, contact rdavidson@uspharmacist.com.



