US Pharm. 2024;49(7):17-28.
ABSTRACT: Cystic fibrosis (CF) is a common genetic disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, resulting in altered function of anion channels in epithelial cells. Multiple organs are involved in CF, notably the lungs and pancreas. The development of CFTR modulator therapies has increased the life expectancy of CF patients. Other CF treatments include pancreatic enzyme replacement therapy for pancreatic insufficiency, mucolytic agents for airway clearance, and systemic and inhaled antibiotics for respiratory infections. Pharmacists can impact CF care through drug management, patient and provider education, and vaccination.
Cystic fibrosis (CF) is the most common autosomal recessive genetic disease and affects approximately 89,000 people in the world.1,2 CF is more common among white non-Hispanic people, and in the United States and Europe the prevalence of CF is almost eight per 100,000 people.2 CF appears to be less common in other parts of the globe, likely due to a lack of prenatal and newborn screening and underreporting.3,4 CF used to be a childhood disease, with most patients dying at a young age; however, with the advent of routine screening and new target CF therapies, life expectancy has increased.4 Most infants born with CF today can be expected to survive into their 50s.2,4
PATHOPHYSIOLOGY
CF is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. These mutations alter anion channels in epithelial cells, resulting in fewer or a complete absence of the channels or decreased channel function.1,2 CFTR mutations are typically divided into six classes. In Classes I, II, and III, the anion channel is not present, is not functional, or is defective with minimal function, resulting in more severe disease. The anion channels are present in Classes IV, V, and VI but have reduced numbers or decreased function. Since residual CFTR function remains in Class IV, V, and VI variants, the disease is usually milder.2,5,6 Class II is the most common variant in the U.S. (85.5% of patients with CF), and the most common mutation is F508del.2
Organ destruction and fibrosis occur due to obstruction of exocrine ducts and altered hydration from reduced channel function.1,2 Since CFTR is found in multiple organ systems, a wide range of conditions can be seen in patients with CF. Patients with CFTR mutations can present with pancreatic insufficiency with malnutrition, biliary liver disease, male infertility, intestinal obstruction and other gastrointestinal problems, delayed development, and respiratory conditions (e.g., sinusitis and bronchitis).1,2
Respiratory disease and bacterial infections occur commonly in CF patients.2 Airway infections with Staphylococcus aureus, Haemophilus influenza, and Pseudomonas aeruginosa can result in bronchiectasis due to the inflammatory response and blockage of airways. Patients can have acute pulmonary exacerbations with cough, increased sputum production, and dyspnea that results in hospitalization and airway-clearance treatment. Chronic endobronchial infections and inflammation lead to obstructive lung disease, with decreased forced expiratory volume in 1 second (FEV1) and forced vital capacity (FVC) on spirometry.2
Exocrine pancreatic insufficiency results in malabsorption of fat, protein, and fat-soluble vitamins (A, D, E, and K). This leads to poor general health, slow growth in children, and weight loss in adults.7 Malnutrition in CF patients increases the risk of respiratory infection and severity of lung disease.7,8 Pancreatic insufficiency can present with flatulence, dyspepsia, bloating, unexplained weight loss or failure to thrive, and loose, foul-smelling, oily stools (steatorrhea). Some patients may be asymptomatic.5
With the progression of CF, endocrine pancreatic tissue is destroyed, and 20% of adolescents and 50% of adults will develop CF-related diabetes.5 Therefore, patients with CF should be screened annually for glucose tolerance. If CF-related diabetes develops, insulin is the treatment of choice. Limited evidence is available for the use of oral hypoglycemic agents, incretins, or incretin-mimetic agents for CF-related diabetes.8
NUTRITION THERAPY
Pancreatic Enzyme Replacement Therapy
The majority of patients with CF are on pancreatic enzyme replacement therapy (PERT) with pancrelipase.7 PERT can prevent malnutrition, promote weight gain, and help control bowel movements. All U.S. pancrelipase formulations are derived from porcine pancreases, although some nonanimal-derived formulations are under investigation.7 Pancrelipase is available as a tablet or a delayed-release capsule.9 The delayed-release capsules contain enteric-coated microspheres that dissolve in the duodenum (where endogenous pancreatic enzymes work) at a pH of 5.0 to 5.5. The enteric coating ensures that the enzymes are not destroyed by gastric acid.5 The tablets do not have an enteric coating and are not delayed-release. Therefore, tablets should be taken with a proton pump inhibitor to prevent breakdown in the stomach.8,9
Dietary Recommendations
Because malnutrition and undernutrition are common in patients with CF, dietary management is vital.8 Nutritional status should be monitored regularly. Dietary counseling, ideally from a dietitian familiar with CF, should be offered to patients, parents, and caregivers. Newly diagnosed infants should be exclusively breastfed, if possible. Breastfed infants with CF have better lung function and fewer infections than infants who are not breastfed. If breastfeeding is not possible or supplementation with formula is necessary, regular infant formula should be used. Evidence does not support the routine use of high-energy or hydrolyzed formula in CF without other reasons. Infants may require sodium supplementation due to sodium loss in sweat.8
Children and adults with CF have increased energy and protein requirements.8 Sodium supplementation may be needed during periods of hot weather or fever or when exercising. Calcium may be low due to vitamin D deficiency, increasing the risk of osteopenia or osteoporosis. Calcium levels should be monitored annually, along with iron and zinc levels; these minerals should be supplemented as needed.8
Since pancreatic insufficiency can decrease the absorption of fat-soluble vitamins, levels of vitamins A, D, and E should be monitored regularly, at least once a year.7,8 Vitamin supplements should be taken with high-fat food and pancreatic enzyme supplements. Vitamin D levels may be adequate in patients with adequate sun exposure. Vitamin K is usually low in CF patients, especially those with CF-related liver disease. Infants with CF who are exclusively breastfed should receive vitamin K supplementation, as well as patients on broad-spectrum antibiotics, those with liver disease, and those with severe malabsorption. Water-soluble vitamin deficiency is rare in uncomplicated CF, but supplementation may be warranted in some patients.8
PHARMACOLOGIC TREATMENT OF CF
Medication management in CF includes the use of CFTR modulators, mucolytics, bronchodilators, and antibiotics to improve mucus clearance and treat infection. Since the guideline update in 2013, additional CFTR modulators have received FDA approval and continue to be a focus of research and development in CF management.
CFTR Modulators
CFTR modulators are classified as potentiators, correctors, stabilizers, amplifiers, or read-through agents based on their effect on the CFTR mutation.6 Currently, FDA-approved therapies for CF are either potentiators or correctors. Potentiators are primarily associated with Class III and IV CFTR variants, most commonly G551D.10 Potentiators enhance CFTR function via channel gating augmentation, promoting mucus transport, and decreasing mucus adhesion to epithelial surfaces.6,11 Correctors are primarily associated with Class II CFTR variants, most commonly F508del, and increase CFTR quantity at the cell membrane.10 Correctors rescue protein folding, processing, and trafficking to a CFTR mutant and enhance protein stability.6
In 2013, the CF Pulmonary Guidelines included the first-approved CFTR modulator therapy, ivacaftor.12 Subsequently, three additional CFTR modulators were approved by the FDA, prompting a guideline update focusing on CFTR modulators in 2018.13 Currently, there are four FDA-approved CFTR modulator therapies, including ivacaftor, lumacaftor/ivacaftor, tezacaftor/ivacaftor, and elexacaftor/tezacaftor/ivacaftor (ETI; see TABLE 1).6,14-18 CFTR modulator selection is based on patient age, CFTR genotype, and clinical or in vitro data supporting CFTR modulator responsiveness.
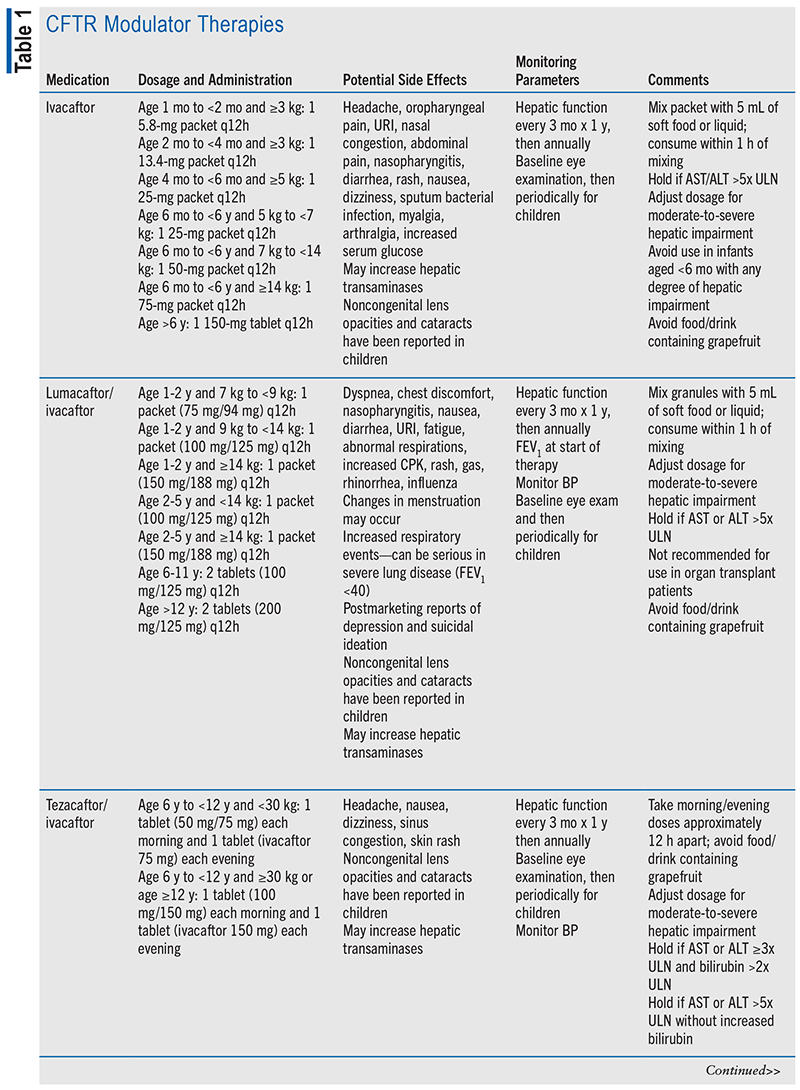
Ivacaftor, a potentiator, is indicated for patients aged 1 month or older with at least one ivacaftor-responsive mutation.15,19 Ivacaftor increases chloride transport via potentiation of CFTR channel gating of the CFTR protein on epithelial cell surfaces.15 Initial clinical trials found that for patients aged 12 years and older with at least one G551D mutation, the change in predicted FEV1 was higher by 10.6 percentage points and individuals were 55% less likely to have pulmonary exacerbations (P <.001). Additionally, body weight increased, overall symptoms decreased, and sweat chloride concentration decreased.20 Further studies in younger participants found similar results.21
Lumacaftor/ivacaftor, a corrector/potentiator combination, is indicated for patients aged 12 months or older with a homozygous F508del mutation. While ivacaftor improves chloride transport via channelgating potentiation, lumacaftor (a corrector) increases the processing and trafficking of mature F508del-CFTR protein to bronchial cell surfaces. This combination therapy improves overall chloride transport, resulting in increased predicted FEV1, decreased pulmonary exacerbations, improved lung clearance index, and reduced sweat chloride concentrations.16,22-24
Tezacaftor/ivacaftor is approved for individuals aged 6 years and older with a homozygous F508del mutation or one tezacaftor/ivacaftor-responsive CFTR mutation.17 This corrector (tezacaftor) and potentiator (ivacaftor) combination increases the quantity and function of cell surface CFTR.17,19 Tezacaftor/ivacaftor increased predicted FEV1 by 4 percentage points (P <.001) and decreased pulmonary exacerbations by 35% (P = .005) in a phase III trial of 510 participants aged 12 years and older.25
ETI is approved for individuals aged 2 years and older with at least one F508del mutation or an ETI-responsive mutation.18 This triple therapy combines one potentiator (ivacaftor) with two correctors (elexacaftor and tezacaftor) to increase CFTR protein quantity and function. By binding two different CFTR-protein sites, elexacaftor and tezacaftor increase cellular processing and trafficking of mutated CFTR proteins. The combination of two different correctors increases the amount of cell surface CFTR protein compared with either corrector alone. The addition of ivacaftor further increases protein function via channel gating potentiation. The results of a phase III, randomized, double-blind, placebo-controlled trial in participants aged 12 years and older (N = 403) found an increase in the percentage of predicted FEV1 of 13.8 points and 14.3 points at Weeks 4 and 24, respectively. Pulmonary exacerbation rates were 63% lower, respiratory symptoms decreased, and sweat chloride concentrations decreased.26 Reductions in sputum production and mucus plugging have also been found.27
While the true impact of CFTR modulators has not been evaluated, the Cystic Fibrosis Foundation (CFF) reports that more than 24,000 individuals with CF were taking a CFTR modulator by the end of 2022, with an estimated 72% taking ETI.28,29 A 2023 systematic review (N = 34 studies) evaluated the effect of CFTR correctors (e.g., elexacaftor, tezacaftor) with or without a potentiator (e.g., ivacaftor) on clinically important benefits in patients with Class II CFTR mutations (most often F508del) in CF patients of any age. Evidence was not sufficient for the use of corrector monotherapy in homozygous F508del mutations. Corrector/potentiator dual therapy resulted in small improvements in quality of life (QoL) and respiratory function and a reduced frequency of pulmonary exacerbations. Triple therapy with two correctors and one potentiator improved QoL and lung function.30
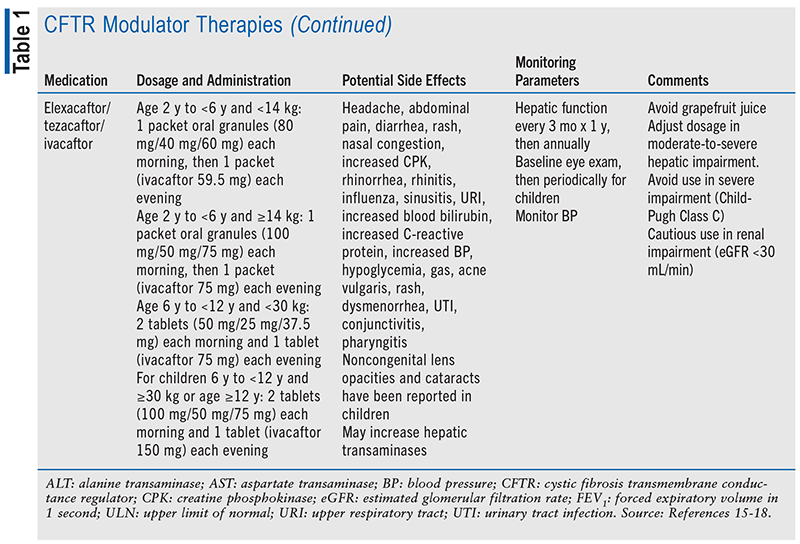
Dosing, potential side effects, and monitoring parameters of CFTR modulators are summarized in TABLE 1. CFTR modulators should be taken with a fat-containing meal to increase absorption. Notably, many potential drug-drug interactions exist and may require dose adjustments or alterations in therapy, especially when used in conjunction with narrow therapeutic index drugs (e.g., warfarin, digoxin).15-18 Potential drug-drug interactions are presented in TABLE 2.
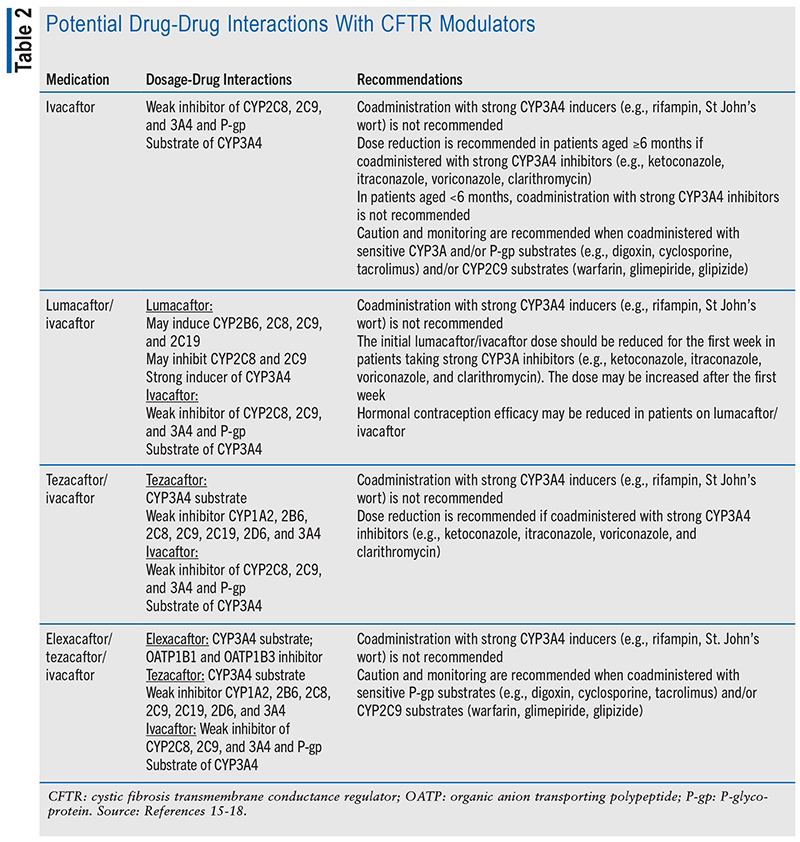
Mucolytic Therapies
Dornase alpha, hypertonic saline, and inhaled mannitol are nebulized therapies that promote airway clearance due to their effects on mucus and sputum (see TABLE 3). Dornase alpha and hypertonic saline are recommended for chronic use to improve lung function and reduce pulmonary exacerbations per the 2013 CFF guidelines.12
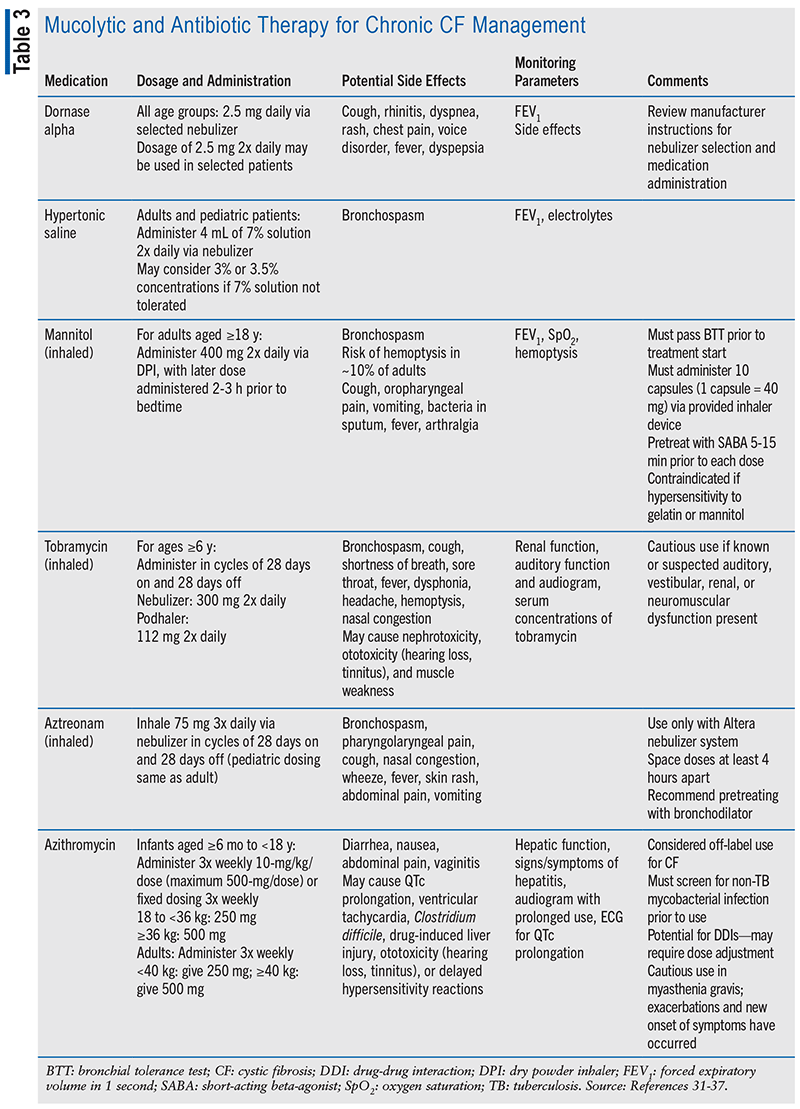
Dornase alpha is indicated for CF management in patients aged 3 months and older. Dornase alpha is a recombinant human deoxyribonuclease I (rhDNase) enzyme that selectively cleaves DNA, resulting in hydrolysis of DNA in the sputum. This allows for increased sputum clearance due to reduced viscosity.31
Nebulized hypertonic saline in concentrations of 3% or 7% hydrates mucus, increases mucociliary clearance, and may reduce inflammatory airway processes.38 Hypertonic saline is recommended for individuals aged 6 years and older to improve lung function and QoL and decrease pulmonary exacerbations. In individuals aged younger than 6 years, hypertonic saline is recommended based on individual circumstances (e.g., pulmonary exacerbation) or if the individual is symptomatic.39,40
Inhaled mannitol may be used as an alternative mucolytic to hypertonic saline in CF patients aged 18 years and older. How inhaled mannitol improves lung function in CF is not known. A systematic review (N = 6 studies) identified that some improvement in lung function measures was seen with inhaled mannitol; however, more research is needed.41 One limiting factor of inhaled mannitol use is the requirement of passing a bronchial tolerance test (BTT) before use due to the risk of severe bronchospasm. The BTT must be administered under the supervision of a healthcare provider.
The combination of CFTR modulators with mucolytics has been evaluated. The SIMPLIFY study evaluated the impact of discontinuing nebulized hypertonic saline or dornase alpha in individuals aged 12 years and older with mild-to-no lung disease treated for 90 days or longer with ETI and either or both hypertonic saline or dornase alpha via two noninferiority studies. SIMPLIFY found that at 6 weeks, lung function was similar whether the mucolytic was stopped or continued.42
Antibiotic Therapy
The most common pathogen found in the airways of individuals with CF is P aeruginosa. Infection with P aeruginosa has been associated with a quicker loss of lung function and reduced survival. For those infected, the 8-year risk of death was 2.6 times higher than in those without P aeruginosa.12,43 P aeruginosa prevalence is decreasing, with 13.5% positive cultures in 2022 versus 43.8% positive cultures in 2002 in adult patients with CF.29 Prevalence is higher in adults than in children. Multidrug-resistant P aeruginosa remains a concern and is more common in older adolescents and adults.29 S aureus is another common pathogen, with methicillin-susceptible S aureus affecting 50.2% of individuals who provided a respiratory culture in 2022. S aureus is more prevalent in younger persons with CF than in older persons.29
Preventative therapy with antibiotics is recommended for individuals with CF aged 6 years and older with persistent presence of P aeruginosa in airway cultures. Inhaled tobramycin, inhaled aztreonam, and oral azithromycin are recommended and have been shown to improve lung function and reduce pulmonary exacerbations (see TABLE 3).12
Inhaled aztreonam prolonged the time to pulmonary exacerbation and decreased the number of hospital days compared with placebo.12,44-46 Azithromycin, in addition to antipseudomonal activity, exerts an anti-inflammatory effect in alveolar macrophages and CF-airway epithelial cells via reduction of proinflammatory mediators.47-49 For patients with sputum-positive nontuberculous mycobacterial infection, azithromycin should not be initiated to reduce the risk of developing macrolide-resistant mycobacterial species. Patients should be screened before initiating chronic azithromycin use and reassessed every 6 to 12 months.12
A systematic review (N = 18 trials) evaluated the effects of long-term inhaled antibiotic therapy on lung function and exacerbation frequency in CF individuals.50 Inhaled antibiotic therapy improved lung function, decreased exacerbations, and resulted in fewer missed days of work/school. While inhaled aztreonam potentially improves lung function and may lead to fewer courses of antibiotics than tobramycin, further research is recommended in this area.50
Ibuprofen
To slow the loss of lung function, chronic use of high-dose ibuprofen is recommended for individuals aged 6 to 17 years with an FEV1 of 60% or more predicted. Ibuprofen dosing is targeted to achieve a peak plasma level of 50 mcg/mL to 100 mcg/mL.12,51 How ibuprofen reduces the progression of lung function loss is not known. It is hypothesized that as a nonsteroidal anti-inflammatory drug, it decreases lung function due to inflammatory responses in CF, and the progression of Pseudomonas-related lung disease is delayed with high-dose ibuprofen use.51 For adults aged 18 years and older, chronic use of high-dose ibuprofen is not recommended due to lack of evidence.
Additional Therapies
Leukotriene modifiers, oral N-acetylcysteine, inhaled glutathione, inhaled anticholinergic bronchodilators, inhaled beta2-agonist bronchodilators, oral antipseudomonal antibiotics, oral antistaphylococcal antibiotics, and other inhaled antibiotics are not recommended for chronic use in CF due to lack of sufficient evidence. The CFF recommends against the use of inhaled and oral corticosteroids and the prophylactic use of oral antistaphylococcal antibiotics.12
ROLE OF THE PHARMACIST
In 2022, approximately 52% of individuals with CF met with a pharmacist at least once.29 Pharmacy-based programs have demonstrated the value of pharmacist inclusion within the CF healthcare team.52,53 CF is a complex health condition with a high medication burden. Pharmacists may assist in medication selection and optimization to reduce treatment burden, improve adherence, decrease medication errors, and reduce cost. Pharmacists may assist with achieving appropriate dosing parameters, evaluating regimens for potential drug-drug or drug-disease interactions, and improving medication access. Education of providers regarding new and emerging pharmacotherapy in CF management and patient self-management education regarding medication use and adherence are other opportunities for pharmacist involvement. Additionally, pharmacy-led immunization programs provide an opportunity to prevent common respiratory illnesses and increase vaccination access. The role of pharmacists in CF management continues to grow, and many opportunities exist to assist in the care of CF patients.
REFERENCES
1. Polgreen PM, Comellas AP. Clinical phenotypes of cystic fibrosis carriers. Annu Rev Med. 2022;73:563-574.
2. Ong T, Ramsey BW. Cystic fibrosis: a review. JAMA. 2023;329(21):1859-1871.
3. Scotet V, L’Hostis C, Férec C. The changing epidemiology of cystic fibrosis: incidence, survival and impact of the CFTR gene discovery. Genes (Basel). 2020;11(6):589.
4. Burgel PR, Burnet E, Regard L, Martin C. The changing epidemiology of cystic fibrosis: the implications for adult care. Chest. 2023;163(1):89-99.
5. Singh VK, Schwarzenberg SJ. Pancreatic insufficiency in cystic fibrosis. J Cyst Fibros. 2017;16(Suppl 2):s70-s78.
6. Lopes-Pacheco M. CFTR modulators: the changing face of cystic fibrosis in the era of precision medicine. Front Pharmacol. 2020;10:1662.
7. Somaraju URR, Solis-Moya A. Pancreatic enzyme replacement therapy for people with cystic fibrosis. Cochrane Database Syst Rev. 2020;8(8):CD008227.
8. Turck D, Braegger CP, Colombo C, et al. ESPEN-ESPGHAN-ECFS guidelines on nutrition care for infants, children, and adults with cystic fibrosis. Clin Nutr. 2016;35(3):557-577.
9. Facts and Comparisons: Drug Referential Resource. Hudson, Ohio: Wolters Kluwer; 2024. www.wolterskluwer.com/en/solutions/uptodate/enterprise/lexidrug-facts-and-comparisons. Accessed April 29, 2024.
10. Southern KW, Castellani C, Lammertyn E, et al. Standards of care for CFTR variant-specific therapy (including modulators) for people with cystic fibrosis. J Cyst Fibros. 2023;22(1):17-30.
11. Birket SE, Chu KK, Houser GH. Combination therapy with cystic fibrosis transmembrane conductance regulator modulators augment the airway function microanatomy. Am J Physiol Lung Cell Mol Physiol. 2016;310:L928-L939.
12. Mogayzel PJ Jr, Naureckas ET, Robinson KA, et al. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med. 2013;187(7):680-689.
13. Ren CL, Morgan RL, Oermann C, et al. Cystic Fibrosis Foundation pulmonary guidelines: use of cystic fibrosis transmembrane conductance regulator modulator therapy in patients with cystic fibrosis. Ann Am Thorac Soc. 2018;15(3):271-280.
14. Taylor-Cousar JL, Robinson PD, Shteinberg M, Downey DG. CFTR modulator therapy: transforming the landscape of clinical care in cystic fibrosis. Lancet. 2023;402:1171-1184.
15. Kalydeco (ivacaftor) product information. Boston, MA: Vertex Pharmaceuticals, Inc; August 2023.
16. Orkambi (lumacaftor/ivacaftor) product information. Boston, MA: Vertex Pharmaceuticals, Inc; August 2023.
17. Symdeko (tezacaftor/ivacaftor) product information. Boston, MA: Vertex Pharmaceuticals, Inc; August 2023.
18. Trikafta (elexacaftor, tezacaftor, and ivacaftor) product information. Boston, MA: Vertex Pharmaceuticals, Inc; August 2023.
19. Goetz DM, Savant AP. Review of CFTR modulators 2020. Pediatr Pulmonol. 2021;56:3595-3606.
20. Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiatior in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663-1672.
21. Davies JC, Wainwright CE, Canny GJ, et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med. 2013;187:1219-1225.
22. Wainright CE, Elborn JS, Ramsey BW, et al. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373:220-231.
23. Ratjen F, Hug C, Marigowda G, et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR: a randomised, placebo-controlled phase 3 trial. Lancet Respir Med. 2017;5:557-567.
24. Milla CE, Ratjen F, Marigowda G, et al. Lumacaftor/ivacaftor in patients aged 6-11 years with cystic fibrosis and homozygous for F508del-CFTR. Am J Respir Crit Care Med. 2017;195:912-920.
25. Taylor-Cousar JL, Munck A, McKone EF, et al. Tezacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N Engl J Med. 2017;377:2013-2023.
26. Middleton PG, Mall MA, Drevinek P, et al. Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med. 2019;381:1809-1819.
27. Bec R, Reynaud-Gaubert M, Arnaud F, et al. Chest computed tomography improvement in patients with cystic fibrosis treated with elexacaftor-tezacaftor-ivacaftor: early report. Eur J Radiol. 2022;154:110421.
28. Cystic Fibrosis Foundation. 2022 Cystic Fibrosis Foundation patient registry highlights. www.cff.org/medical-professionals/patient-registry. Accessed June 10, 2024.
29. Cystic Fibrosis Foundation. 2022 Patient registry annual data report. www.cff.org/medical-professionals/patient-registry. Accessed June 10, 2024.
30. Heneghan M, Southern KW, Murphy J, et al. Corrector therapies (with or without potentiators) for people with cystic fibrosis with class II CFTR gene variants (most commonly F508del). Cochrane Database Syst Rev. 2023;11(11):CD010966.
31. Pulmozyme (dornase alfa) product information. South San Francisco, CA: Genentech, Inc; February 2024.
32. Sodium chloride. In: Lexicomp Online, Lexi-Drugs Online. Waltham, MA: UpToDate, Inc; May 2, 2024. https://online.lexi.com.
33. Bronchitol (mannitol) product information. Frenchs Forest NSW, Australia: Pharmaxis Ltd; October 2020.
34. Tobi (tobramycin) product information. East Hanover, NJ: Novartis Pharmaceuticals, Inc; February 2023.
35. Tobi Podhaler (tobramycin inhalation powder) product information. San Carlos, CA: Mylan Pharmaceuticals, Inc; February 2023.
36. Cayston (aztreonam for inhalation) product information. Foster City, CA: Gilead Sciences, Inc; November 2019.
37. Azithromycin (systemic). In: Lexicomp Online, Lexi-Drugs Online. Waltham, MA: UpToDate, Inc; May 2, 2024. https://online.lexi.com.
38. Wark P, McDonald VM, Smith S. Nebulised hypertonic saline for cystic fibrosis. Cochrane Database Syst Rev. 2023;6(6):CD001506.
39. Lahiri T, Hempstead SE, Brady C, et al. Clinical practice guidelines from the Cystic Fibrosis Foundation for preschoolers with cystic fibrosis. Pediatrics. 2016;137(4):e20151784.
40. Borowitz D, Robinson KA, Rosenfeld M, et al. Cystic Fibrosis Foundation evidence-based guidelines for the management of infants with cystic fibrosis. J Pediatr. 2008;155:S73-S93.
41. Nevitt SJ, Thornton J, Murray CS, Dwyer T. Inhaled mannitol for cystic fibrosis. Cochrane Database Syst Rev. 2020;5:CD008649.
42. Mayer-Hamblett N, Ratjen F, Russell R, et al. Randomized open-label non-inferiority trials evaluating discontinuation versus continuation of hypertonic saline or dornase alfa in modulator treated people with cystic fibrosis: results from the SIMPLIFY study. Lancet Respir Med. 2023;11(4):329-340.
43. Emerson J, Rosenfeld M, McNamara S, et al. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol. 2022;34:91-100.
44. McCoy KS, Quittner AL, Oermann CM, et al. Inhaled aztreonam lysine for chronic airway Pseudomonas aeruginosa in cystic fibrosis. Am J Respir Crit Care Med. 2008;178:921-928.
45. Retsch-Bogart GZ, Quittner AL, Gibson RL, et al. Efficacy and safety of inhaled aztreonam lysine for airway Pseudomonas in cystic fibrosis. Chest. 2009;135:1223-1232.
46. Wainright CE, Quittner AL, Geller DE, et al. Aztreonam for inhalation solution (AZLI) in patients with cystic fibrosis, mild lung impairment, and P. aeruginosa. J Cyst Fibros. 2011;10:234-242.
47. Meyer M, Huaux F, Gavilanes X, et al. Azithromycin reduces exaggerated cytokine production by M1 alveolar macrophages in cystic fibrosis. Am J Respir Cell Mol Biol. 2009;41:590-602.
48. Cigana C, Nicolis E, Pasetto M, et al. Anti-inflammatory effects of azithromycin in cystic fibrosis airway epithelial cells. Biochem Biophys Res Commun. 2006;350:977-982.
49. Wagner T, Soong G, Sokol S, et al. Effects of azithromycin on clinical isolates of Pseudomonas aeruginosa from cystic fibrosis patients. Chest. 2005;128:912-919.
50. Smith S, Rowbotham NJ. Inhaled anti-pseudomonal antibiotics for long-term therapy in cystic fibrosis. Cochrane Database Syst Rev. 2022: 11(11):CD001021.
51. Lands LC, Stanojevic S. Oral non-steroidal anti-inflammatory drug therapy for lung disease in cystic fibrosis. Cochrane Database Syst Rev. 2019;4:CD001505.
52. Abraham O, Li JS, Monangai KE, et al. The pharmacist’s role in supporting people living with cystic fibrosis. J Am Pharm Assoc. 2018;58:246-249.
53. Kim AS, Thompson AN, Axon DR, et al. A scoping review of clinical pharmacist and pharmacy technician contributions to cystic fibrosis care. J Am Coll Clin Pharm. 2023;6(12):1357-1365.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
To comment on this article, contact rdavidson@uspharmacist.com.


