US Pharm. 2016;41(10):HS8-HS14.
New molecular entities (NMEs), as defined by the FDA, are new drug products containing as their active ingredient a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in late 2015 (TABLE 1) detail the basic clinical and pharmacologic profiles of each new drug, as well as key precautions and warnings. Also included is a brief summary of selected pharmacokinetic, adverse-reaction, drug-interaction, and dosing data submitted to the FDA in support of the manufacturer’s New Drug Application. The information for each NME was obtained primarily from sources published prior to FDA approval. Experience clearly demonstrates that many aspects of a new drug’s therapeutic profile are not detected in premarketing studies and emerge after the drug is used in large numbers of patients. For example, “new” adverse reactions for many NMEs often emerge within 2 to 3 years after they first become available. Some of these drugs may eventually acquire at least one black box warning for serious adverse drug reactions or are withdrawn from the market for safety reasons that were not recognized at the time of approval. Hence, while this review offers a starting point for learning about new drugs, it is essential that practitioners be aware of changes in a drug’s therapeutic profile as reported in the pharmaceutical literature and by their own patients.
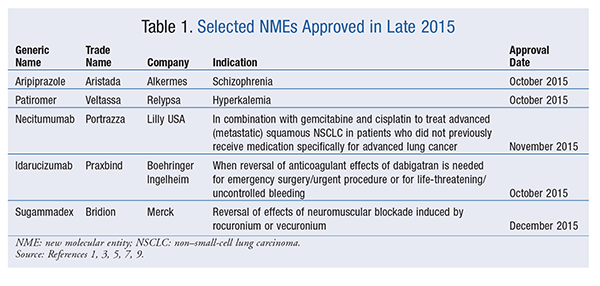
Aripiprazole Lauroxil (Aristada, Alkermes)
Indication and Clinical Profile1,2: Aristada is a long-acting injectable formulation of the antipsychotic aripiprazole that has been approved for the treatment of schizophrenia in patients with established tolerability to oral aripiprazole. Annually, schizophrenia affects about 1% of the U.S. population aged ≥18 years. The disorder is characterized by three symptom types: positive (abnormal behavior), negative (inability to perform daily activities), and cognitive (inability to make decisions).
FDA approval was based on efficacy data from trials with oral aripiprazole as well as a 12-week, controlled, fixed-dose study of 622 adult patients with schizophrenia who had established tolerability to oral aripiprazole. Patients were randomized to Aristada 441 mg (n = 207), Aristada 882 mg (n = 208), or placebo (n = 207) on days 1, 29, and 57. The study’s primary and secondary efficacy outcomes were, respectively, change in Positive and Negative Syndrome Scale (PANSS) score from baseline and improvement based on the Clinical Global Impression Improvement (CGI-I) scale. Patients receiving Aristada 441 mg or 882 mg experienced statistically significant reductions in total PANSS score versus placebo (–10.9 and –11.9, respectively), as well as statistically significantly better CGI-I scores versus placebo.
Pharmacology and Pharmacokinetics1,2: Aristada is a prodrug requiring metabolic conversion to the active antipsychotic aripiprazole following IM injection (FIGURE 1). Upon metabolism to aripiprazole, the drug has activity at multiple neurotransmitter receptors, but primarily acts as a partial agonist at dopamine D2 and serotonin (5-HT)1A receptors and as an antagonist 5-HT2A receptors. The precise mechanism by which Aristada elicits its therapeutic effects in schizophrenia is not fully established, but is thought to involve the drug’s action at a combination of these receptors.
Systemic exposure to aripiprazole occurs within 7 days after initial injection, and release continues for 36 days after administration. Aripiprazole and its major metabolite are 99% plasma protein–bound and have extensive extravascular distribution (volume of distribution, 268 L). Conversion of Aristada involves enzyme-mediated hydrolysis to form N-hydroxymethyl-aripiprazole, followed by elimination to form aripiprazole. Aristada has a terminal half-life ranging from 29 to 35 days and is primarily eliminated via hepatic metabolism involving CYP3A4 and CYP2D6.
Adverse Reactions and Drug Interactions1,2: The most common adverse reaction to Aristada in clinical trials was akathisia. All atypical antipsychotics carry a black box warning alerting prescribers to an increased risk of death associated with off-label use to treat behavioral problems in older people with dementia-related psychosis. As with many atypical antipsychotics, Aristada comes with warnings about cerebrovascular adverse effects including stroke, neuroleptic malignant syndrome, tardive dyskinesias, metabolic changes, orthostatic hypotension, hematologic abnormalities, seizures, and the potential for cognitive and motor impairment. Aristada is secreted into breast milk and should be used with caution in pregnant women because neonates exposed to the drug during the third trimester have experienced extrapyramidal and/or withdrawal symptoms.
Because Aristada is a substrate of CYP2D6 and CYP3A4, its coadministration with drugs that affect these enzymes may require specific dosing adjustments detailed in the prescribing information. This includes strong CYP3A4 inhibitors (e.g., clarithromycin, azole antifungals, protease inhibitors, nefazodone), strong CYP3A4 inducers (e.g., rifampin), and strong CYP2D6 inhibitors (e.g., paroxetine, fluoxetine).
Dosage and Administration1,2: Aristada is available as 5 mL prefilled syringe kits containing sterile aqueous suspensions of 441 mg, 662 mg, and 882 mg of the drug. Prior to initiation, patient tolerability of oral aripiprazole must be established. Aristada may be initiated at a dosage of 441 mg, 662 mg, or 882 mg monthly or as 882 mg every 6 weeks. The drug should be given as an IM injection in either the deltoid muscle (441-mg dose only) or the gluteal muscle (all doses) and should be administered only by a healthcare professional. Use should be avoided in pediatric and elderly patients because safety and efficacy have not been established in these populations. Patients known to be poor metabolizers of CYP2D6 require dose reductions of Aristada. No dose adjustments are required in patients with reduced hepatic or renal function.
Patiromer (Veltassa, Relypsa)
Indication and Clinical Profile3,4: Patiromer is approved for the treatment of hyperkalemia, a serious condition that can result in dangerous, potentially fatal changes in heart rhythm. Potassium levels are regulated by exchange processes in the renal tubules; when the kidneys are unable to remove enough potassium from the blood, the plasma level of potassium can rise dangerously high. Hyperkalemia typically occurs in patients treated with renin-angiotensin-aldosterone (RAA) system inhibitors with stage 3 or greater chronic kidney disease (CKD) who may also have diabetes, heart failure, or both.
FDA approval of patiromer was based on a two-part randomized study that evaluated the drug in hyperkalemic patients with CKD who were taking stable doses of at least one RAA system inhibitor. Patients with a baseline serum potassium level of 5.1 to 5.5 mEq/L were assigned to an initial patiromer dosage of 8.4 g per day, and those with a baseline serum potassium level of 5.5 to 6.5 mEq/L were assigned to 16.8 g per day (in divided doses). The primary efficacy endpoint was the mean change in serum potassium from baseline to week 4. Patients who had a baseline potassium level of 5.5 to <6.5 mmol/L that decreased to 3.8 to <5.1 mmol/L by the end of week 4 entered an 8-week randomized withdrawal phase in which they were randomly continued on patiromer or switched to placebo. The primary efficacy endpoint was the between-group difference in the median change in serum potassium over the first 4 weeks of that phase. In the initial treatment phase, the mean ± SE change in serum potassium in 237 patiromer patients was −1.01 ± 0.03 mmol/L (P <.001). At week 4, 76% (95% CI, 70-81) of patients had reached the target potassium level (3.8-<5.1 mmol/L). Subsequently, 107 patients were randomly assigned to patiromer (n = 55) or placebo (n = 52) for the randomized withdrawal phase. The median increase in potassium from baseline of that phase was greater with placebo than with patiromer (P <.001); a recurrence of hyperkalemia (potassium level ≥5.5 mmol/L) occurred in 60% of placebo patients versus 15% of patiromer patients through week 8 (P <.001).
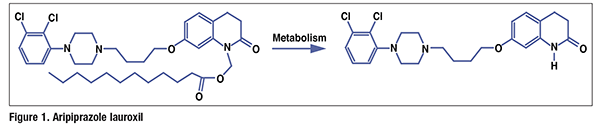
Pharmacology and Pharmacokinetics3,4: Patiromer is a cross-linked polymer of 2-fluoroacrylic acid (91%) with divinylbenzenes (8%) and 1,7-octadiene (1%). It is a calcium salt with sorbitol (one molecule per two calcium ions or four fluoroacrylic acid units), a combination called patiromer sorbitex calcium (FIGURE 2). Patiromer binds free potassium in the gastrointestinal tract and releases calcium ions in exchange, thereby lowering the amount of potassium available for absorption into the systemic circulation and increasing the amount that is excreted fecally. Lowering of potassium levels is detectable 7 hours after administration. Potassium levels continue to decrease for ≥48 hours if treatment is continued, and they remain stable for 24 hours after administration of the last dose. After this, potassium levels start to rise again over a period of ≥4 days. Patiromer is not absorbed from the gut, is not metabolized, and is excreted in unchanged form in the feces.
Adverse Reactions and Drug Interactions3,4: Patiromer was generally well tolerated in clinical trials. Adverse effects occurring in >2% of patients included constipation, diarrhea, nausea, abdominal discomfort, flatulence, and hypomagnesemia (because patiromer also binds magnesium in the gut). Patiromer should not be used as an emergency treatment for life-threatening hyperkalemia because of its delayed onset of action. The drug must be dispensed with a patient medication guide that describes important information about its uses and risks. The drug should be avoided in patients with severe constipation, bowel obstruction, or impaction, including abnormal postoperative bowel-motility disorders, because it may be ineffective and may worsen gastrointestinal conditions. Patiromer is contraindicated in patients with a history of hypersensitivity reaction to the drug or any of its components.
To date, no formal drug-interaction studies of patiromer have been conducted. However, since this drug binds many substances besides potassium, it could reduce the bioavailability of other orally administered drugs given concurrently, thereby reducing their effectiveness. For that reason, patiromer carries a black box warning advising patients to wait ≥6 hours after taking patiromer before taking other oral medications.
Dosage and Administration3,4: Patiromer is supplied as powder for oral suspension in packets containing 8.4 g, 16.8 g, and 25.2 g of the active drug. The powder should be suspended in water immediately before administration. The recommended starting dose is 8.4 g once daily. Serum potassium must be monitored and the dose increased or decreased to reach the desired serum potassium concentration, up to a maximum of 25.2 g once daily. Based on serum potassium levels, the dose may be uptitrated in increments of 8.4 g at intervals of 1 week or more.
Necitumumab (Portrazza, Lilly USA)
Indication and Clinical Profile5,6: Necitumumab has been approved, in combination with gemcitabine and cisplatin, for use in patients with advanced (metastatic) squamous non–small-cell lung cancer (NSCLC) who have not previously received medication specifically for treating advanced lung cancer. Lung cancer is the leading cause of cancer death in the United States, with an estimated 221,200 new diagnoses and 158,040 deaths in 2015. NSCLC, the most common type of lung cancer, is further divided into two main types named for the kinds of cells found in the cancer—squamous cell and nonsquamous cell (which includes adenocarcinoma). Since lung cancer tumors can vary, treatment must be tailored to the patient’s specific type of lung cancer. Necitumumab gives certain NSCLC patients a new option that may extend survival.
FDA approval was based on SQUIRE, a multicenter phase III trial that examined first-line treatment with necitumumab plus gemcitabine and cisplatin versus treatment with gemcitabine and cisplatin alone in 1,093 patients with metastatic squamous NSCLC. Patients in both study arms could receive a maximum of six cycles of chemotherapy, but necitumumab patients who demonstrated at least stable disease continued to receive additional necitumumab cycles until disease progression or unacceptable toxicity. Necitumumab combination therapy resulted in a statistically significant improvement in the main outcome measure of overall survival (OS; P = .01), with a median OS of 11.5 months for the necitumumab arm versus 9.9 months for those treated with gemcitabine and cisplatin alone—a 16% reduction in risk of death. The significant survival improvement observed in SQUIRE was supported by a statistically significant improvement in progression-free survival (PFS; P = .02), with a median PFS of 5.7 months for the necitumumab arm versus 5.5 months for those treated with gemcitabine and cisplatin alone. There was no difference in overall response rate (ORR) between arms, with an ORR of 31% for the necitumumab plus gemcitabine and cisplatin arm and an ORR of 29% for the gemcitabine and cisplatin arm (P = .40). The drug was counterproductive in nonsquamous NSCLC.
Pharmacology and Pharmacokinetics5,6: Necitumumab is a recombinant human immunoglobulin G1 monoclonal antibody that functions as an epidermal growth factor receptor (EGFR) antagonist. Expression and activation of EGFR correlates with malignant progression, induction of angiogenesis, and inhibition of apoptosis. Binding of necitumumab induces EGFR internalization and degradation in vitro and leads to antibody-dependent cellular cytotoxicity (ADCC) in EGFR-expressing cells.
In clinical trials, necitumumab exhibited dose-dependent kinetics, with a mean total systemic clearance of 14 mL per hour, a steady-state volume of distribution of 7.0 L, and an elimination half-life of 14 days. The predicted time to reach steady state is 100 days. In trials, no significant differences in pharmacokinetics were observed in patients with renal or hepatic impairment.
Adverse Reactions and Drug Interactions5,6: The most common adverse reactions to necitumumab in trials were skin rash and hypomagnesemia, which can cause muscle weakness, seizure, and irregular heartbeat and can be fatal. Necitumumab has a black box warning regarding the risk of cardiopulmonary arrest and hypomagnesemia. The label also includes precautions for venous and arterial events, infusion-related and dermatologic toxicities, and embryo-fetal toxicity. The drug should not be used during pregnancy or while breastfeeding. Few drug-interaction studies have been published to date. Necitumumab increases the AUC of gemcitabine by approximately 20% when the drugs are used concurrently, but it has no effect on cisplatin exposure. Neither gemcitabine nor cisplatin alters necitumumab exposure.
Dosage and Administration5,6: Necitumumab is supplied as an 800 mg/50 mL (16 mg/mL) solution in a single-dose vial for IV administration. The recommended dosage is 800 mg given as an IV infusion over 60 minutes on days 1 and 8 of each 3-week cycle prior to gemcitabine and cisplatin infusion. Patients who experienced grade 1 or 2 infusion-related reactions upon previous administration should be pretreated with antihistamines (diphenhydramine) prior to subsequent doses. Specific dose modifications for patients who experience infusion or dermatologic toxicities are provided in the manufacturer’s literature. Therapy should continue until disease progression or unacceptable toxicity develops.
Idarucizumab (Praxbind, Boehringer Ingelheim)
Indication and Clinical Profile7,8: Idarucizumab has been approved for use in patients treated with the anticoagulant dabigatran when it is necessary to reverse dabigatran’s blood-thinning effects for an emergency surgery or urgent procedure or for life-threatening or uncontrolled bleeding. Dabigatran has advantages over warfarin as an anticoagulant because it causes fewer drug interactions and has a reduced need for monitoring. However, the inability to reverse the actions of dabigatran when needed is a significant disadvantage. Because of its efficacy and large potential benefit, idarucizumab was approved under the FDA’s accelerated-approval program, but continued approval is contingent on the results of an ongoing cohort case-series study.
FDA approval of idarucizumab was based on recent results of the ongoing RE-VERSE AD trial. This single-cohort case series studied idarucizumab 5 g in 123 dabigatran-treated patients who either experienced serious bleeding or required an urgent procedure. The primary outcome was the maximum percentage of dabigatran reversal within 4 hours of administration of idarucizumab as measured by dilute thrombin time or ecarin clotting time. At the time of interim analysis, idarucizumab use in both of the patient groups was associated with a median maximum dabigatran reversal of 100% at 4 hours, with >89% of patients achieving complete reversal at that point.
Pharmacology and Pharmacokinetics7,8: Idarucizumab is a humanized monoclonal antibody fragment derived from an immunoglobulin G1 isotype molecule. It binds to dabigatran with a higher binding affinity than the affinity of dabigatran for thrombin, outcompeting thrombin and effectively preventing dabigatran from exerting its pharmacologic effect.
Idarucizumab does not demonstrate any significant extravascular distribution, and its mean volume of distribution is 8.9 L. There are multiple pathways of idarucizumab metabolism, all of which are a part of the typical protein catabolic processes and do not involve CYP enzymes. Idarucizumab undergoes rapid clearance at a rate of 47 mL/min, resulting in an initial half-life of 47 minutes and a terminal half-life of 10.3 hours. Although the primary route of idarucizumab elimination is protein catabolism, approximately 33% of the drug is excreted unchanged in the urine.
Adverse Reactions and Drug Interactions7,8: The most common adverse reactions reported by trial patients receiving idarucizumab included hypokalemia, confusion, constipation, fever, and pneumonia. Although idarucizumab does not possess procoagulant effects, dabigatran reversal exposes patients to the risk of blood clots from their underlying disease state. Because idarucizumab contains sorbitol as an excipient, it should be used with caution in patients with hereditary fructose intolerance because it puts these patients at increased risk for fatal adverse reactions such as hypoglycemia, hypophosphatemia, metabolic acidosis, hyperuricemia, and acute liver failure. The potential for idarucizumab to cause fetal harm is unknown; it has not been studied in pregnant humans or animals and should be given to a pregnant woman only if it is clearly needed.
No known drug interactions with idarucizumab have been reported at this time. It is not metabolized by, nor does it have any known effect on, CYP enzymes, and it is not known to have pharmacologic interactions with any drug other than its mechanistic effect on dabigatran. Available data suggest that the therapeutic effect of idarucizumab is not impacted by the presence of coagulation factor concentrates or by hemodilution up to 50% using volume-replacement strategies. Idarucizumab may be used in conjunction with standard all-supportive measures.
Dosage and Administration7,8: Idarucizumab is supplied in single-use vials of 2.5 g/50 mL solution for IV injection. The recommended dosage is 5 g either as two consecutive infusions or by bolus injection of both vials. Currently, data supporting the administration of an additional 5 g are limited. Idarucizumab should not be administered simultaneously with other infusions at the same IV access, and when a preexisting line is used to administer idarucizumab, the line must be flushed with sterile 0.9% sodium chloride. Dabigatran therapy may be reinitiated 24 hours after idarucizumab administration, and it should be resumed as soon as appropriate. Idarucizumab does not require dose adjustments in elderly patients or those with renal impairment. Its safety and efficacy have yet to be established in pediatric patients or those with hepatic impairment.
Sugammadex (Bridion, Merck)
Indication and Clinical Profile9,10: Sugammadex is approved by the FDA to reverse the effects of neuromuscular blockade induced by rocuronium and vecuronium. Rocuronium and vecuronium are aminosteroidal neuromuscular-blocking agents (NMBAs) that are used for tracheal intubation in adults. They also may be used to prevent patient movement while under general anesthesia and sometimes may be used to prevent spontaneous breathing in ventilated patients. Rocuronium and vecuronium have an intermediate duration of action.
The efficacy of sugammadex was investigated in three phase III clinical trials including 456 patients who underwent elective surgical procedures under general anesthesia. The return to recovery time was faster overall in the sugammadex treatment groups versus the comparator groups, and most participants recovered within 5 minutes of routine use of sugammadex. In another trial involving 189 patients, sugammadex was compared with neostigmine for reversal of the neuromuscular blockade induced by rocuronium and vecuronium during elective surgical procedures. In this study, sugammadex 2 mg/kg resulted in faster recovery from NMBA blockade than did neostigmine 50 mcg/kg. Based on concerns about the nature and frequency of potentially life-threatening anaphylaxis and hypersensitivity reactions reported in the clinical trials, sugammadex was further evaluated in a randomized, parallel-group, repeat-dose trial. Of the 299 participants treated with sugammadex, one person had an anaphylactic reaction. The drug has not been studied for NMBA reversal after administration of rocuronium or vecuronium in the ICU, and it should not be used to reverse the blockade induced by nonsteroidal NMBAs (e.g., succinylcholine) or by steroidal NMBAs other than rocuronium or vecuronium.
Pharmacology and Pharmacokinetics9,10: The mechanism of action of sugammadex differs significantly from that of other commonly used reversal agents, such as neostigmine and edrophonium. Sugammadex is a modified gamma-cyclodextrin with a lipophilic core and a hydrophilic periphery. This gamma-cyclodextrin has been modified from its natural state by the substitution of eight carboxyl thioether groups at the sixth carbon positions (FIGURE 3). These negatively charged extensions electrostatically bind to the quaternary nitrogen of the target NMBA, extend the cavity size (which allows for greater encapsulation of the rocuronium molecule), and enhance the aqueous nature of the cyclodextrin. The high-affinity binding of rocuronium by sugammadex renders the NMBA unable to bind to the nicotinic receptor at the neuromuscular junction. Sugammadex also has affinity for two other aminosteroid neuromuscular blocking agents: vecuronium and pancuronium. Although sugammadex has a lower affinity for vecuronium than for rocuronium, vecuronium reversal is effective because it is approximately seven times more potent than rocuronium, and therefore lower doses are used. Shallow pancuronium blockade has been successfully reversed by sugammadex in phase III clinical trials.
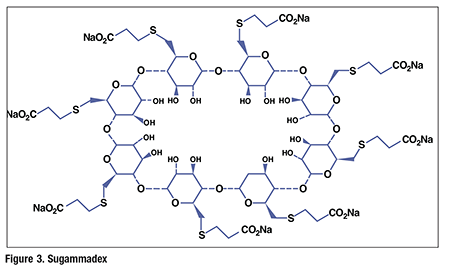
The steady-state volume of distribution of sugammadex is about 11 to 14 L in patients with normal renal function. Sugammadex and the complex of sugammadex-rocuronium are not bound by plasma proteins or erythrocytes. Sugammadex is retained in sites of active mineralization, such as bone and teeth, with a mean half-life of 172 and 8 days, respectively. Sugammadex is not metabolized, and it is excreted exclusively by the kidneys at an elimination half-life of about 2 hours. Therefore, this drug is not recommended for use in patients with severe renal impairment, including those requiring dialysis.
Adverse Reactions and Drug Interactions9,10: The most common adverse reactions reported in clinical trials of sugammadex included vomiting, hypotension, pain, headache, and nausea. Cases of marked bradycardia, some of which have resulted in cardiac arrest, have been observed within minutes of administration of the drug; therefore, patients should be closely monitored for hemodynamic changes during and after reversal of neuromuscular blockade, and treatment with anticholinergic agents such as atropine should be administered if clinically significant bradycardia is observed. Potentially serious hypersensitivity reactions, such as anaphylaxis, have occurred in sugammadex-treated patients, and known hypersensitivity to sugammadex is a contraindication to use of the drug. Clinicians should be aware of the possibility of a hypersensitivity reaction or anaphylaxis and should intervene as appropriate.
Sugammadex can bind to progestogen and may temporarily reduce its contraceptive effect. Therefore, female patients who have received sugammadex and are currently using hormonal contraceptives should be advised to also use a nonhormonal method of contraception for the next 7 days. No other clinically significant drug interactions are anticipated.
Dosage and Administration9,10: Bridion is supplied in single-dose vials containing 2 or 5 mL of sugammadex 100 mg/mL, and the vials should be stored at 25°C and protected from light. The labeling recommends a single 2- to 4 mg/kg bolus injection of the reversal agent, with the timing and dose dependent on the patient’s response to train-of-four stimulation. A 16 mg/kg bolus injection is an option only if the patient received a single 1.2 mg/kg dose of rocuronium, and the clinical need for reversal of neuromuscular blockade arises within about 3 minutes after NMBA administration. All recommended dosages of sugammadex are based on actual body weight.
REFERENCES
1. Aristada (aripiprazole lauroxil) package insert. Waltham, MA: Alkermes, Inc; October 2015.
2. Citrome L. Aripiprazole long-acting injectable formulations for schizophrenia: aripiprazole monohydrate and aripiprazole lauroxil. Expert Rev Clin Pharmacol. 2016;9:169-186.
3. Veltassa (patiromer) package insert. Redwood City, CA: Relypsa, Inc; October 2015.
4. Henneman A, Guirguis E, Grace Y, et al. Emerging therapies for the management of chronic hyperkalemia in the ambulatory care setting. Am J Health Syst Pharm. 2016;73:33-44.
5. Portrazza (necitumumab) package insert. Indianapolis, IN: Lilly USA, LLC; November 2015.
6. Thatcher N, Hirsch FR, Luft AV, et al; SQUIRE Investigators. Necitumumab plus gemcitabine and cisplatin versus gemcitabine and cisplatin alone as first-line therapy in patients with stage IV squamous non-small-cell lung cancer (SQUIRE): an open-label, randomised, controlled phase 3 trial. Lancet Oncol. 2015;16:763-774.
7. Praxbind (idarucizumab) package insert. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals, Inc; October 2015.
8. Pollack CV Jr, Reilly PA, Eikelboom J, et al. Idarucizumab for dabigatran reversal. N Engl J Med. 2015;373:511-520.
9. Bridion (sugammadex) package insert. Whitehouse Station, NJ: Merck & Co, Inc; December 2015.
10. Pühringer FK, Rex C, Sielenkämper AW, et al. Reversal of profound, high-dose rocuronium-induced neuromuscular blockade by sugammadex at two different time points: an international, multicenter, randomized, dose-finding, safety assessor-blinded, phase II trial. Anesthesiology. 2008;109:188-197.
To comment on this article, contact rdavidson@uspharmacist.com



