US Pharm. 2019;44(10):HS6-HS12.
New molecular entities (NMEs), as defined by the FDA, are new drug products containing as their active ingredient a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in 2018–2019 (TABLE 1) detail the basic clinical and pharmacologic profiles of each new drug, as well as key precautions and warnings. Also included is a brief summary of selected pharmacokinetic, adverse-reaction, drug-interaction, and dosing data submitted to the FDA in support of the manufacturer’s New Drug Application. The information for each NME was obtained primarily from sources published prior to FDA approval. Experience clearly demonstrates that many aspects of a new drug’s therapeutic profile are not detected in premarketing studies and emerge after the drug is used in large numbers of patients. For example, “new” adverse reactions for many NMEs often emerge within 2 to 3 years after they first become available. Some of these drugs may eventually acquire at least one black box warning for serious adverse drug reactions or are withdrawn from the market for safety reasons that were not recognized at the time of approval. Therefore, while this review offers a starting point for learning about new drugs, it is essential that practitioners be aware of changes in a drug’s therapeutic profile as reported in the pharmaceutical literature and by their own patients.
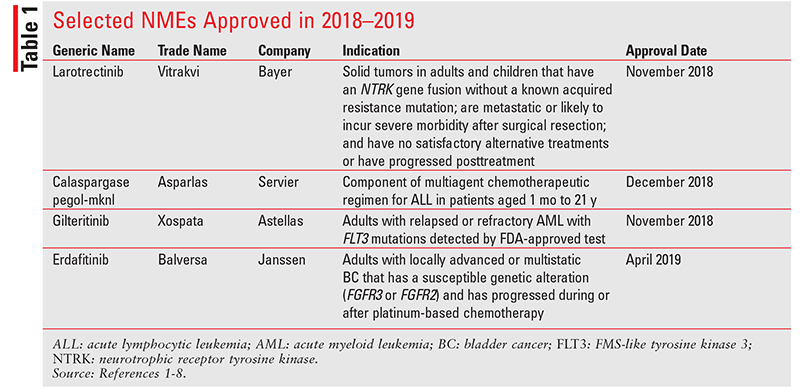
Larotrectinib (Vitrakvi, Bayer)
Indication and Clinical Profile1,2: In November 2018, the FDA granted larotrectinib accelerated approval and orphan-drug designation for treatment of adults and children with solid tumors that have a neurotrophic receptor tyrosine kinase (NTRK) gene fusion without a known acquired resistance mutation; are metastatic or likely to incur severe morbidity after surgical resection; and have no satisfactory alternative treatments or have progressed posttreatment. This approval marks a new paradigm in the development of cancer drugs designed to treat any cancer that expresses certain mutations (tissue-agnostic cancer drugs) rather than cancers of specific tissues. Larotrectinib was initially awarded orphan-drug status in 2015 for soft-tissue sarcoma, and it received a breakthrough designation in 2016 for treatment of metastatic solid tumors with an NTRK gene fusion. Prior to the November 2018 approval, there was no treatment for cancers that frequently express this mutation, such as mammary analogue secretory carcinoma, cellular or mixed congenital mesoblastic nephroma, and infantile fibrosarcoma.
FDA approval was based on the results of pooled data from three clinical trials involving 55 pediatric and adult patients who had solid metastatic tumors with an identified NTRK gene fusion and no resistance mutation. Patients either had no satisfactory alternative treatments or had cancer progression following treatment. Larotrectinib demonstrated a 75% overall response rate across different types of solid tumors. Responses were durable, with 73% lasting at least 6 months and 39% lasting 1 year or longer at the time that results were analyzed. Median time to response was 1.84 months. Progression-free survival was not reached at the time of analysis. Tumor types with an NTRK fusion that responded to larotrectinib therapy included soft-tissue sarcoma, salivary-gland cancer, infantile fibrosarcoma, thyroid cancer, and lung cancer.
Pharmacology and Pharmacokinetics1,2: Larotrectinib (FIGURE 1) is a pyrazolopyrimidine that functions as a highly selective inhibitor of the tropomyosin receptor kinases (TRKs) TRKA, TRKB, and TRKC. TRKA, TRKB, and TRKC are encoded by the genes NTRK1, NTRK2, and NTRK3; chromosomal rearrangements involving in-frame fusions of these genes with various partners may result in constitutively activated chimeric TRK fusion proteins that can act as an oncogenic driver, promoting cell proliferation and survival in tumor cell lines. In tumor models, larotrectinib demonstrated antitumor activity in cells by activating TRK proteins that result from gene fusions or deletion of a protein regulatory domain, and in cells with TRK overexpression.
Larotrectinib has 34% oral bioavailability. Standard doses produce a peak plasma concentration of 788 ng/mL within 1 hour and achieve steady state within 3 days. In healthy subjects, peak plasma concentration was 36% higher with oral solution than with capsules. The drug has a volume of distribution of 48 L and is 70% bound by plasma proteins. Larotrectinib is metabolized predominantly by CYP3A4 and has an elimination half-life of 2.9 hours, with a clearance of 98 L/hour, and it is excreted in the feces (58%, 5% unchanged) and urine (39%, 20% unchanged).
Adverse Reactions and Drug Interactions1,2: Common adverse effects (>20%) reported in clinical trials included fatigue, nausea, cough, constipation, diarrhea, dizziness, vomiting, and increased aspartate aminotransferase (AST) and alanine aminotransferase (ALT) enzyme levels. The majority of neurologic adverse reactions occurred within the first 3 months of treatment, and patients should be advised not to drive or operate hazardous machinery if they are experiencing neurologic effects. Patients should have ALT and AST liver tests every 2 weeks during the first month of treatment, then monthly and as clinically indicated. Women who are pregnant or breastfeeding should not take larotrectinib because it may cause harm to the fetus or newborn. Females and males should be advised to use effective contraception during treatment and for at least 1 week after the last dose. Larotrectinib is a CYP3A4 substrate and inhibitor; therefore, coadministration with strong CYP3A4 inhibitors, strong CYP3A4 inducers, and sensitive CYP3A4 substrates should be avoided.
Dosage and Administration1,2: Larotrectinib is supplied as both a capsule (25 and 100 mg) and an oral solution (20 mg/mL). The recommended dosage for adult and pediatric patients with a body surface area of at least 1.0 m2 is 100 mg orally twice daily, and pediatric patients with body surface area less than 1.0 m2 should receive 100 mg/m2 orally twice daily. The capsules should be swallowed whole with water, not chewed or crushed. The capsules and oral solution may be used interchangeably. If vomiting occurs, the next dose should be taken at the scheduled time. Missed doses (within 6 hours of next scheduled dose) should not be made up.
No dosage adjustments are required for mild-to-severe renal impairment or mild (Child-Pugh A) hepatic impairment. In moderate-to-severe (Child-Pugh B-C) hepatic impairment, the larotrectinib dose should be reduced by 50%. The drug may be taken with or without food and may be continued until the occurrence of disease progression or unacceptable toxicity. If any grade ≥3 adverse reactions occur, treatment should be withheld until the reaction resolves or improves to grade 1 or lower and resumed at the next recommended dose modification if resolved within 4 weeks. If the reaction does not resolve within 4 weeks, the drug should be permanently discontinued.
The use of strong CYP3A4 inhibitors should be avoided. If coadministration cannot be avoided, the larotrectinib dose should be reduced by 50%. Once a strong CYP3A4 inhibitor is discontinued for three to five elimination half-lives, administration may be resumed at the dose taken prior to CYP3A4 inhibitor initiation. The use of strong CYP3A4 inducers should also be avoided. If coadministration cannot be avoided, the larotrectinib dose should be doubled. Once the strong CYP3A4 inducer is discontinued for three to five elimination half-lives, administration may be resumed at the dose taken prior to initiation of the CYP3A4 inducer. Concurrent use of larotrectinib and sensitive CYP3A4 substrates should be avoided. If coadministration cannot be avoided, the patient should be monitored for increased CYP3A4 substrate–related adverse reactions.
Calaspargase pegol-mknl (Asparlas, Servier)
Indication and Clinical Profile3,4: Calaspargase pegol-mknl, an asparagine-specific enzyme, is approved as a component of a multiagent chemotherapeutic regimen for treatment of acute lymphoblastic leukemia (ALL) in patients aged 1 month to 21 years. This agent permits a longer interval between doses compared with other available pegaspargase products.
FDA approval was based on a study that demonstrated the achievement and maintenance of nadir serum asparaginase activity (NSAA) >0.1 U/mL with calaspargase pegol-mknl at a dosage of 2,500 U/m2 IV every 3 weeks. Calaspargase pegol-mknl was given in combination with multiagent chemotherapy to 124 patients with B-cell lineage ALL, and the drug’s pharmacokinetics was assessed. In the study, 123 patients (99%) maintained NSAA >0.1 U/mL at weeks 6, 12, 18, 24, and 30. Calaspargase pegol-mknl received FDA orphan-product designation.
In another trial, 237 patients with newly diagnosed ALL or lymphoblastic lymphoma received calaspargase pegol-mknl 2,500 U/m2 (n = 118) or pegaspargase at 2,500 U/m2 (n = 119) as part of Dana-Farber Cancer Institute ALL Consortium backbone therapy. Patients’ median age was 5 years (range, 1-20 years), 62% were male, and 70% were Caucasian. Fifty-nine percent of patients had standard-risk disease, and 87% had B-cell lineage ALL. A median of 11 doses of calaspargase pegol-mknl (every 3 weeks) or 16 doses of pegaspargase (every 2 weeks) was administered. The median duration of treatment exposure was 8 months in both arms. In the subgroup of patients with B-cell lineage ALL, the complete remission rate in the calaspargase pegol-mknl arm was 98% versus 99% in the pegaspargase arm. Additionally, Kaplan-Meier overall survival estimates were comparable between arms.
Pharmacology and Pharmacokinetics3,4: Calaspargase pegol-mknl is an engineered protein consisting of Escherichia coli–derived enzyme L-asparaginase II conjugated with succinimidyl carbonate monomethoxypolyethylene glycol. The L-asparaginase portion of the drug hydrolyzes L-asparagine to L-aspartic acid and ammonia, thereby depriving tumor cells of the L-asparagine they need for growth and replication. Leukemic cells with low expression of asparagine synthetase have a reduced ability to synthesize L-asparagine; therefore, they depend on an exogenous source of L-asparagine for survival. Conjugation with the pegol group increases the drug’s half-life, making it longer-acting. With standard doses, peak plasma concentrations are achieved within 1.2 hours and reach 1.62 U/mL. The drug’s volume of distribution is nearly 3 L, and it has an AUC0-infinity of 25.5 day • U/mL. Calaspargase pegol-mknl has an elimination half-life of 16 days and a clearance of 0.147 L/day.
Adverse Reactions and Drug Interactions3,4: The most common (incidence ≥10%) grade ≥3 adverse reactions reported in clinical trials included elevated transaminase, increased bilirubin, pancreatitis, and abnormal clotting. Accordingly, patients should be carefully monitored during therapy. Less common adverse effects (<10%) included diarrhea, embolic and thrombotic events, sepsis, dyspnea, hemorrhage, fungal infection, pneumonia, arrhythmia, and cardiac failure. In a comparative trial, the safety profile of calaspargase pegol-mknl administered every 3 weeks was similar to that of pegaspargase administered every 2 weeks.
Based on studies in pregnant animals, calaspargase pegol-mknl can cause fetal harm in pregnant women. There are no data on the presence of calaspargase pegol-mknl in human milk, the effects on the breastfed child, or the effects on milk production. Because many drugs are excreted in human milk and owing to the potential for adverse reactions in a child who is breastfed, lactating women should be advised not to breastfeed while receiving calaspargase pegol-mknl and for 3 months after the last dose.
No drug-interaction studies have been reported.
Dosage and Administration3,4: Calaspargase pegol-mknl is supplied as single-dose vials (3,750 U/5 mL, 750 U/mL) of solution for IV administration. The recommended dosage is 2,500 U/m2 IV no more frequently than every 21 days. Pregnancy testing should be conducted in females of reproductive potential before treatment is initiated. Patients should be monitored at least weekly, including bilirubin, transaminases, glucose, and clinical examinations, until recovery from the therapy cycle.
If a grade 1 infusion or hypersensitivity reaction occurs, the rate of infusion should be reduced by 50%. For grade 2 infusion or hypersensitivity reactions, the infusion should be interrupted, symptoms treated until resolved, and the infusion resumed at a 50% reduced rate. For grade 3-4 infusion or hypersensitivity reactions, the drug should be discontinued permanently. For grade 3-4 hemorrhage, administration should be withheld, and the patient should be evaluated for coagulopathy and considered for clotting factor replacement. Administration may be resumed at next scheduled dose if bleeding is controlled. For grade 3-4 pancreatitis, administration should be withheld for elevated lipase or amylase (>3 × upper limit of normal [ULN]) until enzymes levels stabilize or are declining, and the drug should be discontinued permanently if clinical pancreatitis is confirmed. In the case of uncomplicated deep venous thrombosis, administration should be withheld and the patient treated with appropriate antithrombotic therapy. Administration may be resumed upon resolution of symptoms while antithrombotic therapy is continued. The drug should be discontinued in the case of severe or life-threatening thrombosis. In cases of hepatotoxicity, if the total bilirubin is >3 to ≤0 × ULN, administration should be withheld until total bilirubin decreases to ≤1.5 × ULN. If the total bilirubin is >10 × ULN, the drug should be discontinued.
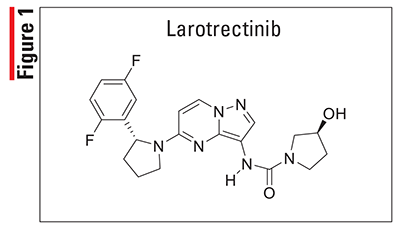
Gilteritinib (Xospata, Astellas)
Indication and Clinical Profile5,6: Gilteritinib is specifically indicated for treatment of adults who have relapsed or refractory acute myeloid leukemia (AML) with an FMS-like tyrosine kinase 3 (FLT3) mutation detected by an FDA-approved test. AML is a rapidly progressing cancer in which the bone marrow makes abnormal myeloblasts, RBCs, or platelets, resulting in low numbers of normal blood cells and, consequently, a need for continuous transfusions and treatments to avoid anemia, bleeding, or an immunocompromised state. According to the National Cancer Institute, more than 61,000 people had a diagnosis of AML in 2016, and it was estimated that 21,450 people would be diagnosed with AML and 10,920 AML-related deaths would occur in 2019. AML from an FLT3 gene mutation is an aggressive form of disease that has a higher rate of relapse and is present in roughly 25% to 30% of patients with AML. Gilteritinib is the first approved drug that may be used alone to treat patients with AML with an FLT3 mutation who have relapsed or do not respond to initial treatment.
FDA approval was based on data from a multicenter, randomized, open-label, parallel-assignment trial that included 138 subjects who had relapsed or refractory AML with an FLT3 mutation. Participants received gilteritinib 120 mg daily until occurrence of unacceptable toxicity or a lack of clinical benefit. Endpoints included the rate of complete remission/complete remission with partial hematologic recovery (CR/CRh); duration of CR/CRh; and rate of conversion from transfusion dependence to transfusion independence. Upon study completion, 21% of gilteritinib patients achieved CR/CRh (CR, 11.6%; CRh, 9.4%), and 31% of patients who required RBC or platelet transfusions at treatment initiation were transfusion-free for at least 56 days.
Pharmacology and Pharmacokinetics5,6: Gilteritinib (FIGURE 2) is a 2-pyrazinecarboxamide that inhibits multiple receptor tyrosine kinases, including FLT3. In normal cells, the binding of FLT3 and its cytokine ligand results in signaling cascades that promote the transcription of genes regulating survival, proliferation, and differentiation. Certain FLT3 mutations that lead to AML result in FLT3 that uses ligand-independent signaling, which promotes cytokine-independent AML-cell survival and proliferation. Through its effects on FLT3, gilteritinib inhibits FLT3 receptor signaling and proliferation of cells exogenously expressing FLT3, including those with internal tandem duplications (ITD) and the tyrosine kinase domain mutations FLT3-D835Y and FLT3-ITD-D835Y, and it induces apoptosis in leukemic cells expressing FLT3-ITD.
Gilteritinib achieves its peak plasma concentration 4 to 6 hours post administration, and steady-state plasma levels are reached within 15 days of administration. It is 94% plasma protein bound and is extensively distributed to tissue, with a central and peripheral volume of distribution of 1,092 L and 1,100 L, respectively. Gilteritinib is primarily metabolized via N-dealkylation by CYP3A4 to three primary metabolites, none of which exceeds 10% of overall gilteritinib exposure. The drug has an elimination half-life of 113 hours and is primarily excreted in the feces (64.5%), with 16.4% excreted in the urine.
Adverse Reactions and Drug Interactions5,6: The most common (≥20%) adverse reactions reported in clinical trials were edema, hypotension, fatigue, malaise, headache, dizziness, skin rash, hypocalcemia, hypoalbuminemia, hypophosphatemia, hypokalemia, hyponatremia, diarrhea, constipation, nausea, vomiting, stomatitis, arthralgia, myalgia, dyspnea, pneumonia, cough, and fever, as well as elevations in serum glucose, triglycerides, hepatic enzymes, creatine phosphokinase, and creatinine. Patients receiving gilteritinib may experience differentiation syndrome, which can be fatal if untreated. Patients suspected to have differentiation syndrome should be treated with routine corticosteroids for a minimum of 3 days, and interruption of gilteritinib is required if severe signs or symptoms continue for more than 48 hours after corticosteroid initiation. The drug should be discontinued in patients who experience confirmed posterior reversible encephalopathy syndrome (PRES) during treatment. Other possible gilteritinib-related toxicities that may require dose modifications include QT-interval prolongation and pancreatitis.
Owing to its CYP3A metabolism, gilteritinib should be used with extreme caution in patients taking strong CYP3A inhibitors (e.g., azole antifungals, protease inhibitors) because these medications will cause an increase in gilteritinib exposure. Concomitant use of gilteritinib and a combined P-glycoprotein pump and strong CYP3A inducer (e.g., doxorubicin, phenobarbital, prazosin) should be avoided because these agents reduce exposure of gilteritinib, which may lead to a loss of efficacy.
Dosage and Administration5,6: Gilteritinib is supplied as a 40-mg tablet for oral administration. The recommended starting dosage is 120 mg once daily. ECGs and regular evaluations of patient blood counts and blood chemistries are required to monitor for gilteritinib-related toxicities (e.g., PRES, QTc-interval prolongation) prior to and during treatment because patients experiencing these toxicities may require dose modification or discontinuation of gilteritinib. The safety and efficacy of gilteritinib have not been studied in pediatric patients or in patients with severe renal or hepatic impairment. The drug is contraindicated in patients with a previous serious hypersensitivity reaction to gilteritinib or any of its components.
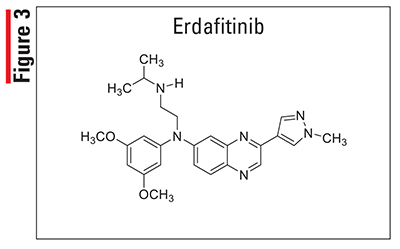
Erdafitinib (Balversa, Janssen)
Indication and Clinical Profile7,8: Bladder cancer is the sixth most common cancer in the United States, and the most common type of bladder cancer is transitional-cell carcinoma (also known as metastatic urothelial carcinoma). Bladder cancers are associated with genetic mutations present in the patient’s bladder or entire urothelium. Fibroblast growth factor receptor (FGFR) alterations occur in approximately 20% of patients with recurrent and refractory bladder cancer. The FDA granted accelerated approval and breakthrough-therapy designation to erdafitinib for treatment of adults who have locally advanced or metastatic bladder cancer with a susceptible FGFR3 or FGFR2 genetic alteration that progressed during or after prior platinum-containing chemotherapy. In selecting patients for erdafitinib therapy, the FDA-approved FGFR RGQ RT-PCR Kit should be used as a companion diagnostic for this therapeutic indication.
Accelerated approval was based on results of a phase II, multicenter, open-label study of 87 patients with bladder cancer that progressed during or after at least one prior chemotherapy. All patients also had at least one FGFR3 gene mutation (R248C, S249C, G370C, Y373C) or FGFR gene fusion (FGFR3-TACC3, FGFR3-BAIAP2L1, FGFR2-BICC1, FGFR2-CASP7) as documented by clinical assay. The overall response rate was 32.2%; 2.3% of patients had a complete response, and almost 30% had a partial response. The responses lasted for an average of 5.4 months. About 25% of patients previously underwent anti–programmed death-ligand 1/programmed cell death protein 1 (PD-L1/PD-1) therapy, which is a standard treatment for patients with locally advanced or metastatic bladder cancer, and responses to erdafitinib were seen in some patients who previously did not respond to anti–PD-L1/PD-1 therapy. Continued approval for this indication will likely be contingent upon verification of clinical benefit in confirmatory trials.
Pharmacology and Pharmacokinetics7,8: Erdafitinib (FIGURE 3) is a pyrazoloquinoxaline that acts like a FGFR kinase inhibitor, inhibiting the enzymatic activity of FGFR1, FGFR2, FGFR3, and FGFR4. Erdafitinib also binds to RET, CSF1R, PDGFRA, PDGFRB, FLT4, KIT, and VEGFR2. Erdafitinib inhibits FGFR phosphorylation and signaling and decreases cell viability in cell lines expressing FGFR genetic alterations, including point mutations, amplifications, and fusions. Erdafitinib demonstrated antitumor activity in FGFR-expressing cell lines and xenograft models derived from a number of tumor types, including bladder cancer.
Erdafitinib is well absorbed after oral administration, reaching peak plasma levels (1,399 ng/mL) at 2.5 hours after standard dosing. Its volume of distribution is 29 L, and it is more than 99% bound by plasma proteins. Erdafitinib is primarily metabolized by CYP2C9 (39%) and CYP3A4 (20%), and patients who have the CYP2C9*3/*3 genotype (0.4%-3% of the population) are predicted to have an approximate 50% increase in systemic erdafitinib exposure and should be monitored for increased adverse reactions. Erdafitinib has an elimination half-life of 59 hours and is excreted in the feces (69%) and urine (19%), primarily as metabolites.
Adverse Reactions and Drug Interactions7,8: Common side effects (>10%) in clinical trials included increased phosphate levels, mouth sores, fatigue, decreased kidney function, diarrhea, dry mouth, nail-structure abnormalities, changes in liver function, low sodium levels, decreased appetite, change in sense of taste, anemia, dry skin, dry eyes, and hair loss. Other side effects included redness, swelling, peeling or tenderness of the hands or feet, constipation, stomach pain, nausea, and muscle pain. Patients receiving this drug should have their blood-phosphate level checked between 14 and 21 days after treatment initiation, and monthly thereafter. Erdafitinib may cause serious eye problems, including inflamed eyes, inflamed cornea, and retinal disorders. Patients are advised to have intermittent eye examinations and to tell their healthcare provider immediately if they develop blurred vision, loss of vision, or other visual changes. Erdafitinib should be withheld if central serous retinopathy or retinal pigment epithelial detachment occurs, and the drug should be permanently discontinued if the condition does not resolve within 4 weeks or is of grade 4 severity.
Pregnant and breastfeeding women should not take erdafitinib because it may cause harm to the fetus or newborn. Pregnancy testing is recommended for females of reproductive potential prior to initiation of erdafitinib treatment. Females of reproductive potential should be advised to use effective contraception during erdafitinib treatment. No data are available on the drug’s presence in human milk, effects on breastfed children, or effects on milk production. Male patients with female partners of reproductive potential should use effective contraception during erdafitinib treatment and for 1 month after the last dose. Erdafitinib must be dispensed with a patient medication guide that describes important information about the drug’s uses and risks.
Based on its pharmacokinetic profile, erdafitinib has a number of significant pharmacologic, metabolic, and transporter-drug interactions. The use of serum phosphate–altering agents should be avoided before the initial dose-modification period because serum phosphate levels are required to determine safety-related dose adjustments. Strong CYP2C9 or CYP3A4 inducers should be avoided, if possible, because they may compromise erdafitinib’s efficacy. If moderate CYP2C9 or CYP3A4 inducers are used concurrently, the erdafitinib dose should be increased to 9 mg. Concurrent use of strong CYP2C9 or CYP3A4 inhibitors may lead to increased drug-related toxicity; therefore, alternative agents should be considered. If coadministration cannot be avoided, the patient should be closely monitored for adverse reactions, and recommended dose modifications should be considered. Concomitant use of sensitive CYP3A4 substrates or those with narrow therapeutic indices should also be avoided because erdafitinib may alter their plasma concentrations, resulting in loss of activity or increased toxicity of CYP3A4 substrates. Erdafitinib administration should occur at least 6 hours before or after administration of P-glycoprotein substrates with narrow therapeutic indices. Coadministration of erdafitinib with OCT2 substrates may increase plasma concentrations of these substrates, resulting in potential toxicity.
Dosage and Administration7,8: Erdafitinib is supplied as 3-, 4-, and 5-mg tablets for oral administration. The recommended starting dosage is 8 mg (two 4-mg tablets) orally once daily. Dose increases are based on serum phosphate levels 14 to 21 days after treatment initiation; the dosage should be increased to 9 mg (three 3-mg tablets) once daily if the serum phosphate level is <5.5 mg/dL, treatment is tolerated, and there are no ocular disorders or grade ≥2 adverse reactions. The tablets should be swallowed whole with or without food. If vomiting occurs at any time after administration, the next dose should be taken the following day. If a dose is missed, it may be taken as soon as possible on the same day and the regular daily dose taken the next day. Extra tablets should not be taken to make up for missed doses. Treatment should continue until disease progression or unacceptable toxicity occurs.
Serum phosphate levels should be monitored monthly for hyperphosphatemia, and phosphate intake should be restricted to 600 to 800 mg per day. If serum phosphate increases to >7 mg/dL, oral phosphate binder therapy should be considered until the phosphate level returns to <5.5 mg/dL.
Management of adverse reactions of erdafitinib, including hyperphosphatemia, may require dose interruption, reduction, or discontinuation depending on the grade of the adverse reaction. The manufacturer’s package insert should be consulted for the management of specific adverse reactions. No dose adjustments are recommended for mild-to-moderate renal impairment (estimated glomerular filtration rate 30-89 mL/min/1.73 m²) or mild hepatic impairment (total bilirubin [TB] ≤ upper limit of normal [ULN] and aspartate aminotransferase [AST] > ULN or TB >1-1.5 × ULN and any AST). Dose recommendations for severe renal impairment and moderate-to-severe hepatic impairment have not been established.
REFERENCES
1. Vitrakvi (larotrectinib) package insert. Whippany, NJ: Bayer HealthCare Pharmaceuticals, Inc; November 2018.
2. Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in TRK fusion–positive cancers in adults and children. N Engl J Med. 2018;378(8):731-739.
3. Asparlas (calaspargase pegol-mknl) package insert. Boston, MA: Servier Pharmaceuticals LLC; December 2018.
4. Vrooman LM, Blonquist TM, Supko JG, et al. Efficacy and toxicity of pegaspargase and calaspargase pegol in childhood acute lymphoblastic leukemia/lymphoma: results of DFCI 11-001. J Clin Oncol. 2019;37(15 suppl):10006-10006.
5. Xospata (gilteritinib) package insert. Northbrook, IL: Astellas Pharma US, Inc; November 2018.
6. Gorcea CM, Burthem J, Tholouli E. ASP2215 in the treatment of relapsed/refractory acute myeloid leukemia with FLT3 mutation: background and design of the ADMIRAL trial. Future Oncol. 2018;14(20):1995-2004.
7. Balversa (erdafitinib) package insert. Horsham, PA: Janssen Products, LP; April 2019.
8. Siefker-Radtke AO, Necchi A, Park SH, et al. First results from the primary analysis population of the phase 2 study of erdafitinib (ERDA; JNJ-42756493) in patients (pts) with metastatic or unresectable urothelial carcinoma (mUC) and FGFR alterations (FGFRalt). J Clin Oncol. 2018;36(suppl); abstract 4503.
To comment on this article, contact rdavidson@uspharmacist.com.



