US Pharm. 2024;49(1):17-26.
ABSTRACT: Historically, the only approach available for treating patients who have genetic deficiencies that cause obesity has been advice to adopt a healthy diet and exercise plan. Setmelanotide is a novel drug therapy for obesity caused by these specific genetic disorders (specifically, Bardet-Biedl syndrome and deficiencies in pro-opiomelanocortin, proprotein convertase subtilisin/kexin type 1, and leptin receptors). Setmelanotide’s mechanism of action targets the hypothalamic melanocortin-4 receptor pathway to control hunger and food intake, leading to weight loss in patients with these rare genetic disorders. Given this agent’s favorable results in clinical trials, it is essential to remember that setmelanotide is indicated only for use in obese patients with these specific genetic disorders.
According to the World Health Organization, the global prevalence of obesity has tripled since 1975, primarily owing to adaptation to a more sedentary lifestyle and the consumption of unhealthy diets.1 In addition, obesity rates have increased to the extent that the obese population has a higher mortality rate than underweight and normal-weight populations. Not only does obesity affect patients’ well-being and quality of life, but it also increases their risk for developing diabetes, cardiovascular diseases (including stroke), and hypertension.1
A sedentary lifestyle and poor diet are not the only contributors to obesity. There are a number of health conditions that disrupt the balance between food intake and energy expenditure, rendering these patients obese. Among these conditions are some rare genetic disorders, such as pro-opiomelanocortin (POMC) deficiency, proprotein convertase subtilisin/kexin type 1 (PCSK1) deficiency, leptin receptor (LEPR) deficiency, and Bardet-Biedl syndrome.
Neurologic Mechanism of Hunger and Satiety
The hypothalamus is the part of the brain that is responsible for regulating the appetite. It does so via neurons clustered in an area called the arcuate nucleus. The arcuate nucleus allows peripheral peptides and hormones to cross the blood-brain barrier and interact directly with neurons to transmit hunger or satiety signals between the stomach and the hypothalamus. The arcuate nucleus contains appetite-stimulating neurons, which express neuropeptide Y and agouti-related peptide (AgRP) and are activated by hunger, and appetite-suppressive neurons, which express the POMC gene and are stimulated by satiety or fullness.2
POMC and PCSK1 Deficiency
The POMC gene is expressed by the pituitary gland, which converts POMC predominantly to adrenocorticotropic hormone (ACTH) via proprotein/prohormone convertase 1 (an enzyme encoded by the PCSK1 gene). ACTH, which is then cleaved to alpha–melanocyte-stimulating hormone (aMSH) via proprotein convertase 2, is mainly involved in skin-color regulation. As shown in FIGURE 1, leptin—a peptide synthesized by adipose tissue—is thought to induce the conversion of POMC to aMSH in the arcuate nucleus of the hypothalamus. Acting as a neurotransmitter, aMSH binds to melanocortin-4 receptors (MC4Rs) in the paraventricular nucleus as well as various sites in the brain. This receptor is heavily involved in the regulation of appetite and energy, as it promotes satiety and energy expenditure when it is bound to aMSH and stimulates hunger and energy conservation when it is bound to AgRP.2
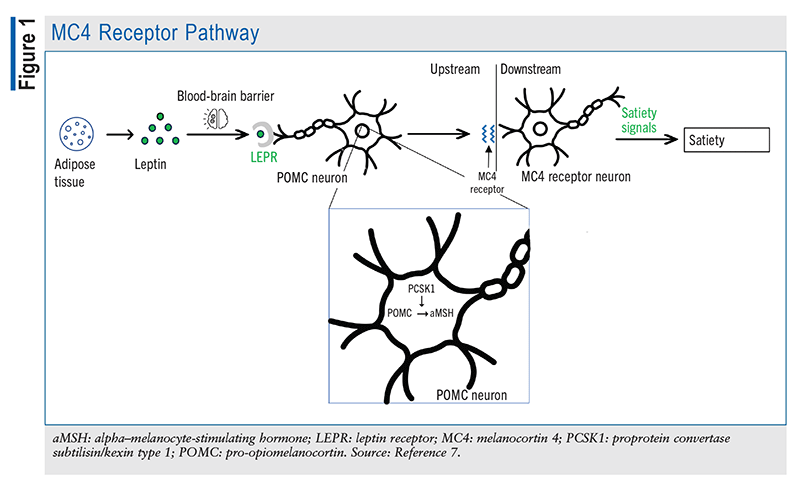
The POMC and PCSK1 genes help maintain homeostasis by effectively delivering signals to the brain to stimulate or inhibit hunger. Accordingly, individuals who are deficient in the genes that help regulate the inhibition of hunger struggle to control their appetite. As depicted in FIGURE 2, POMC-deficient patients are unable to produce sufficient aMSH to bind MC4Rs to stimulate satiety and energy expenditure; additionally, AgRP is constantly binding MC4R to send hunger signals to the brain, with nothing to antagonize its effect. Other symptoms of POMC deficiency include low levels of ACTH and growth hormone, mild hypothyroidism, liver disease, pale skin, and hypogonadotropic hypogonadism resulting in delayed puberty.3
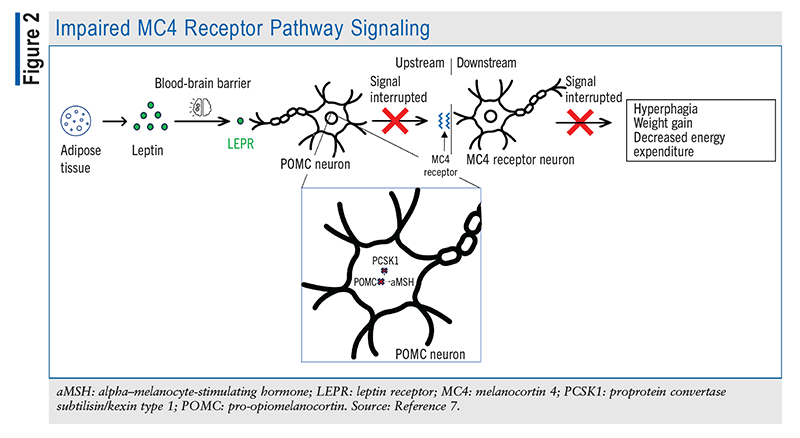
Likewise, PCSK1 gene deficiency translates to a deficiency in the proprotein convertase 1 enzyme, which hinders the normal activity of the converting enzyme. Patients experience intestinal malabsorption for the first few years of life, requiring nutritional therapy. Later on, patients develop hyperphagia and binge eating, resulting in weight gain and obesity that may persist into adulthood.4
A deficiency in the LEPR gene renders the body unable to produce enough LEPRs. Consequently, leptin hormone cannot bind to its receptors to deliver satiety signals to the brain, misleading patients to continue to feel hungry. This circumstance results in the occurrence of early-onset obesity.5
Deficiencies in POMC, PCSK1, and LEPR involve different genes. All of these deficiencies are rare, as they are inherited in an autosomal-recessive manner. Diagnosis requires the assessment of clinical signs and symptoms, along with genetic testing for confirmation.4
Bardet-Biedl Syndrome
Bardet-Biedl syndrome, a rare genetic disorder, has an estimated prevalence of 1,500 to 2,500 persons in the United States, with nine in 10 patients aged 5 years and older being obese. Unlike the three disorders discussed above, Bardet-Biedl syndrome is caused not by a genetic deficiency, but rather by mutations in at least 24 genes that result in defective formation and activity of cilia, which play a vital role in regulating satiety signals. Bardet-Biedl syndrome is part of a group of nonmotile ciliopathies and is inherited in an autosomal-recessive fashion.6
Bardet-Biedl syndrome affects multiple organs and body systems, including the eyes, kidneys, liver, brain, and digestive system. Rod-cone dystrophy affects approximately 90% of Bardet-Biedl syndrome patients, leading to vision impairment or blindness. Early diagnosis of Bardet-Biedl syndrome often is made after the patient develops rod-cone dystrophy, primarily between ages 5 and 10 years. Other symptoms and signs of Bardet-Biedl syndrome include hyperphagia, renal impairment, hypogonadism, and postaxial polydactyly. These clinical signs and symptoms, in addition to genetic testing, are used to diagnose Bardet-Biedl syndrome.7
BBSome is a protein complex responsible for regulating the transport, localization, and signaling of the primary cilia involved in mediating the movement of LEPRs to the surface of POMC neurons via signal transduction. Specifically, the primary cilium present on POMC neurons acts as an antenna that senses the leptin hormone released by the adipose tissue and sends signals inducing LEPR expression on the surface of the POMC neuron; this leads to the binding of leptin to LEPR and the cleavage of POMC, which ultimately activates the MC4R pathway and sends satiety signals. Accordingly, mutations in the genes that encode for BBSome proteins result in Bardet-Biedl syndrome ciliopathy. Patients with Bardet-Biedl syndrome often experience hyperphagia caused by a disruption in the MC4R pathway that leads them to continue to feel hungry despite consuming sufficient food for fullness.8
Treatment
Because of the rare nature of genetic obesity disorders and limited research data, until recently there was no specific drug treatment. Obese patients with these genetic conditions historically have been advised to adopt a healthy diet and an exercise plan to lose weight. However, given that these disorders cause hyperphagia, patients cannot control their appetite without addressing its cause.7
Setmelanotide, a novel drug therapy, is an agonist of the MC4R pathway that is designed to promote chronic weight management in patients aged 6 years and older by increasing satiety and reducing hunger, as shown in FIGURE 3. This agent, which was FDA approved in 2020, is administered as a subcutaneous injection once daily in the morning without regard to meals. Patients should initiate therapy with a 2-mg dose before increasing it to 3 mg after 2 weeks, if tolerated.9 If the 2-mg dose is not tolerated, it is recommended to decrease the dose to 1 mg for 2 weeks and then increase it to 2 mg.9
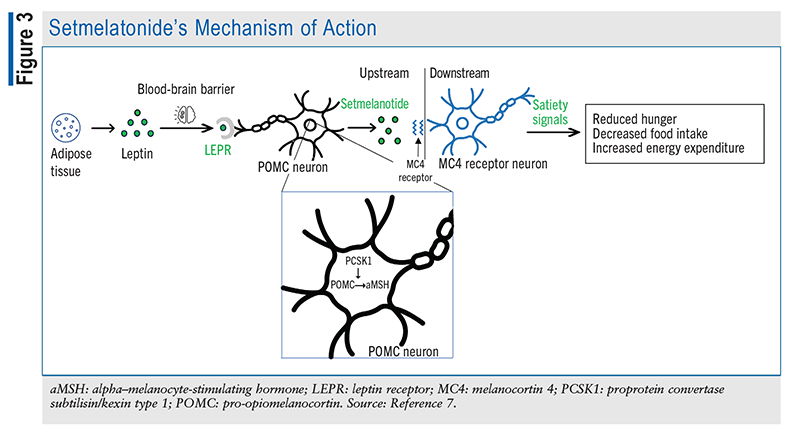
Genetic Considerations for Setmelanotide Use
The specific genetic alterations that result in the rare conditions that setmelanotide is intended to treat involve the MC4R pathway—more specifically, this pathway’s disruption by genetic abnormalities, which is the root cause of the extreme obesity occurring in affected individuals.7 It is essential to remember that setmelanotide is recommended only for patients with certain genetic disorders, including POMC deficiency, PCSK1 deficiency, LEPR insufficiency, and Bardet-Biedl syndrome. There is no universal cure for obesity that is unrelated to these genetic abnormalities.7 The use of setmelanotide should be thoroughly evaluated and recommended by a healthcare specialist, with the decision based on the individual patient’s genetic profile and medical history.7
Safety Considerations With Setmelanotide Use
In clinical trials that assessed the safety of setmelanotide, injection-site reactions, skin hyperpigmentation, nausea, headaches, abdominal pain, diarrhea, and back pain were typical side effects. While taking setmelanotide, some patients may experience an increase in blood pressure. Therefore, the patient’s blood pressure and weight should be routinely monitored at every office visit.7
Setmelanotide’s Efficacy in Clinical Trials
Three separate phase III trials were conducted to assess setmelanotide’s efficacy for three different deficiencies and disorders: Study 1 (POMC or PCSK1 deficiency), Study 2 (LEPR deficiency), and Study 3 (Bardet-Biedl syndrome).10,11 All three trials involved a small number of patients compared with other drug trials because of the rarity of these disorders. Study 1 and Study 2 were conducted over the course of 52 weeks, and Study 3 was conducted over 66 weeks. The efficacy results of each trial are summarized in TABLE 1.10,11
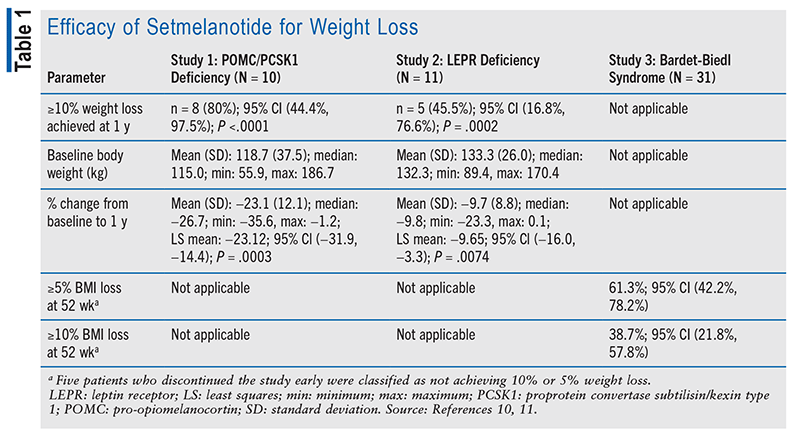
Studies 1 and 2 had similar cohorts and interventions; however, they treated different deficiencies. For both studies, the primary endpoint was a weight loss of at least 10% following 1 year of treatment. Eighty percent of patients in Study 1 and 46% of those in Study 2 achieved a weight reduction of at least 10% after 1 year of treatment. The studies also measured patients’ weight change and hunger scores over the course of 52 weeks. Patients in Study 1 achieved an approximate weight decrease of 24% and a 2-point decrease in hunger score after 1 year of treatment; by contrast, patients in Study 2 experienced an approximate 10% decrease in weight and a 4-point decrease in hunger score following 1 year of treatment.10
Study 3 involved patients aged 6 years and older who were obese and had a clinical diagnosis of Bardet-Biedl syndrome. To be enrolled, adults had to have a BMI of at least 30 kg/m2 and pediatric patients had to be in at least the 97th percentile on the growth-assessment chart. At week 52 of the study, 61% of patients had attained a BMI reduction of at least 5% and 39% of patients had attained a BMI reduction of at least 10%. Over the course of 52 weeks, patients who were receiving setmelanotide treatment had an average reduction of 8% in BMI and 2-point reduction in hunger score.11
Conclusion
Setmelanotide offers hope for patients with POMC deficiency, PCSK1 deficiency, LEPR insufficiency, and Bardet-Biedl syndrome, as it has demonstrated potential success in the treatment of obesity caused by these rare genetic disorders. This agent exerts its action by triggering the hypothalamic MC4R pathway, which controls hunger and energy use, thereby inducing weight loss. Because setmelanotide is intended to treat specific genetic obesity disorders, it should be prescribed and administered only by physicians skilled in treating these conditions. Patients who have Bardet-Biedl syndrome or have deficits in POMC, PCSK1, or LEPR should undergo genetic testing and receive specialized care based on their unique needs. In the public at large, combating the global obesity epidemic calls for comprehensive strategies that include public-health initiatives, education, lifestyle changes, and healthcare regulations that encourage healthier diets and increased physical activity.
REFERENCES
1. World Health Organization. Obesity and overweight. www.who.int/news-room/fact-sheets/detail/obesity-and-overweight. June 9, 2021. Accessed July 10, 2023.
2. Austin J, Marks D. Hormonal regulators of appetite. Int J Pediatr Endocrinol. 2009;2009:141753.
3. Lead for Rare Obesity. Proopiomelanocortin (POMC) deficiency. https://leadforrareobesity.com/pomc-deficiency. Accessed July 19, 2023.
4. Ramos-Molina B, Martin MG, Lindberg I. PCSK1 variants and human obesity. Prog Mol Biol Transl Sci. 2016;140:47-74.
5. Lead for Rare Obesity. Leptin receptor (LEPR) deficiency. https://leadforrareobesity.com/lepr-deficiency. Accessed July 19, 2023.
6. Tsyklauri O, Niederlova V, Forsythe E, et al. Bardet-Biedl syndrome ciliopathy is linked to altered hematopoiesis and dysregulated self-tolerance. EMBO Rep. 2021;22(2):e50785.
7. Imcivree setmelanotide injection [for healthcare providers]. www.imcivree.com/hcp/ppl. Accessed July 20, 2023.
8. Lee CH, Kang GM, Kim MS. Mechanisms of weight control by primary cilia. Mol Cells. 2022;45(4):169-176.
9. Imcivree (setmelanotide) product information. Boston, MA: Rhythm Pharmaceuticals, Inc; November 2023.
10. Clément K, van den Akker E, Argente J, et al. Efficacy and safety of setmelanotide, an MC4R agonist, in individuals with severe obesity due to LEPR or POMC deficiency: single-arm, open-label, multicentre, phase 3 trials. Lancet Diabetes Endocrinol. 2020;8(12):960-970.
11. Haws RM, Gordon G, Han JC, et al. The efficacy and safety of setmelanotide in individuals with Bardet-Biedl syndrome or Alström syndrome: phase 3 trial design. Contemp Clin Trials Commun. 2021;22:100780.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
To comment on this article, contact rdavidson@uspharmacist.com.


