US Pharm. 2018;43(11)(Specialty&Oncology suppl):15-22.
ABSTRACT: Chimeric antigen receptor T (CAR-T) cells represent a novel and personalized therapeutic approach to cancer immunotherapy. CAR-T cells are genetically engineered, autologous T-cells that are modified to create a tumor-specific antigen response. CAR-T cell therapy offers high response rates in patients with relapsed or refractory B-cell malignancies whose treatment options are limited. The hallmark toxicity of CAR-T cell therapy is cytokine release syndrome, an inflammatory response resulting from supraphysiological T-cell activation. Additional toxicities of CAR-T cell therapy include CAR-T cell–related encephalopathy syndrome and B-cell aplasia. The immediate recognition and management of these distinct, potentially life-threatening toxicities is essential to sustaining the unique benefits of CAR-T cell therapy.
The innovation of CD19-directed chimeric antigen receptor T (CAR-T) cells has paved the way for a novel therapeutic approach to the management of some relapsed and refractory (RR) B-cell leukemias and lymphomas. This landmark advancement has demonstrated high response rates in RR acute lymphoblastic leukemia (ALL) and diffuse large B-cell lymphoma (DLBCL).
ALL, a malignant proliferation of lymphoblasts that invade the blood and bone marrow, is the most common childhood leukemia and the second most common acute leukemia in adults.1 Approximately 80% of cases are B-cell ALL; the rest are T-cell ALL. Traditional induction chemotherapy yields initial response rates of up to 80%. However, about 20% of ALL patients fail to respond to initial chemotherapy, another 20% experience relapse, and 5-year overall survival (OS) is as low as 20%, demonstrating an unmet need.2
Non-Hodgkin lymphoma (NHL) is a spectrum of lymphoid malignancies, and in 2018 an estimated 74,680 cases were newly diagnosed in the United States.3 DLBCL, the most common subtype, is aggressive, accounting for approximately 25% of NHL cases. Response rates for initial rituximab-based chemotherapy are high; however, 20% to 50% of patients remain refractory or relapse after primary response.4 Historically, salvage chemotherapy for RR DLBCL fails in 64% to 80% of patients (median OS 6.3 months), and fewer than 10% achieve complete response (CR).4 Old age and comorbidities often preclude candidacy for autologous or allogeneic hematopoietic stem-cell transplantation (HSCT), thereby limiting potentially curative treatment options. The uniformly poor prognosis for RR disease has highlighted the urgent need for more effective therapies, prompting the initiation of CAR-T cell research.
THE CAR-T CELL CONSTRUCT
A fundamental element of the immune response against cancer is T-cell receptor recognition of tumor antigens. When the receptor engages with the target antigen, a downstream signaling cascade of cytokine release and T-cell proliferation occurs in order to eradicate malignant cells. Over time, malignant cells evade T-cell surveillance, and unregulated tumor growth ensues. The goal of CAR-T cell therapy is to restore antitumor immune surveillance by enabling T cells to identify tumor cells independently of the T-cell receptor and major histocompatibility complex–mediated tumor antigen presentation.5
CAR-T cells are autologous T cells genetically engineered to express a surface CAR, which confers new antigen specificity. The CAR is a transmembrane fusion protein with two essential components: an extracellular antigen-recognition domain and an intracellular signaling domain.5 The evolution of CAR-T cell design provides insight into the necessary components of the signaling domain that aids in a potent sustainable immunologic response. Historically, CAR-T cells comprising only a single signaling domain (CD3 zeta) generated insufficient T-cell activation and subsequent limited durability. The addition of a costimulatory domain (CD28 or 4-1BB) confers a robust intracellular-activation signal.5,6 This important contribution has allowed for enhanced T-cell expansion and long-term persistence.6 With guidance from their genetically engineered receptors, these personalized cells create a newly educated immune system to target the recipient’s cancer.
ENGINEERING PROCESS
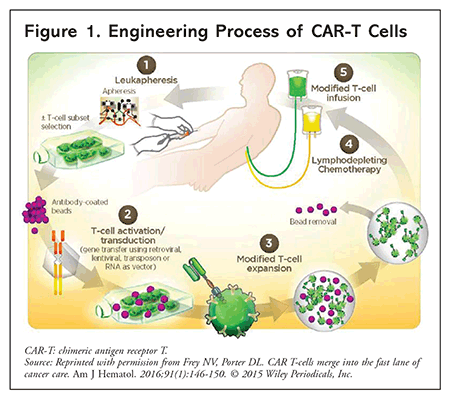
The first step in engineering CAR-T cells is to harvest the recipient’s T cells from whole blood, a process known as leukapheresis. The T cells are then shipped to the manufacturer to be genetically modified with an attenuated lentiviral or retroviral vector, which transfers a specialized DNA sequence encoding the CAR gene. Once created, the cells replicate in vitro, and after cryopreservation, the T-cell product is shipped back to the treatment facility. The entire process takes approximately 2 to 4 weeks.7 In the interim, the patient receives lymphodepleting chemotherapy (often, fludarabine and cyclophosphamide) to provide transient tumor control and to create space for CAR-T cell expansion within the bone marrow. Later, the patient receives CAR-T cells as a single IV infusion via a central line, after which the cells expand in vivo. The recommended time between lymphodepletion and CAR-T cell infusion is 3 days for axicabtagene ciloleucel (axi-cel) and 2 to 14 days for tisagenlecleucel.8,9 FIGURE 1 depicts the engineering process.
CLINICAL-TRIAL DATA
The most extensively investigated CAR-T cell target is the CD19 antigen. CD19 is a B-cell surface protein constitutively expressed on normal and malignant B cells. The lack of expression on hematopoietic stem cells, plasma cells, and nonhematologic tissues minimizes off-target effects, making this an attractive target.10 Tisagenlecleucel and axi-cel are CD19-directed CAR-T cells that have altered the therapeutic landscape for B-cell malignancies.
Tisagenlecleucel (Kymriah, Novartis), the first-in-class CAR-T cell therapy, was approved in August 2017 for RR B-cell ALL in patients aged up to 25 years.8 ELIANA, a phase II, multicenter trial, assessed tisagenlecleucel in 75 patients with RR ALL.11 Patients, who were aged 3 to 23 years (median age 11 years), were heavily pretreated (median three prior therapies; 61% had prior allogeneic HSCT) and had an extensive disease burden (median 74% blasts on bone-marrow biopsy). Median follow-up was 13 months. The observed overall response rate (ORR) was 81%, and OS rates were 90% and 76% at 6 and 12 months, respectively.11
In October 2017, axi-cel (Yescarta, Kite Pharma) became the first CAR-T cell therapy FDA-approved for RR DLBCL after two or more lines of chemotherapy.9 Approval was based on ZUMA-1, a landmark multicenter, phase II trial.12 In the 101 patients treated, an initial ORR of 82% (with 50% CR) was observed. Further follow-up at a median of 15.4 months showed that many responses were durable, with 42% ORR and 40% CR.12 Following this success, based on results of the JULIET trial, tisagenlecleucel received FDA approval for RR DLBCL in May 2018.13 This multicenter, phase II trial evaluated 68 patients who received two or more lines of chemotherapy (median three prior therapies) or relapsed following autologous HSCT. Notably, 50% ORR and 32% CR were attained. Sustained responses beyond 1 year were observed in 83% of patients who achieved a CR at 3 months.13 These groundbreaking clinical trials have expanded the options for treating RR B-cell malignancies.
TOXICITIES
Although CAR-T cell therapy represents a paradigm shift in the armamentarium of B-cell malignancies, the benefits come at the expense of potentially serious adverse effects (AEs). The major treatment-emergent AEs of CAR-T cell therapy are cytokine release syndrome (CRS), CAR-T cell–related encephalopathy syndrome (CRES), and B-cell aplasia.14 Clinician awareness of how to recognize and manage these distinct toxicities is vital to maintaining clinical efficacy while minimizing the risk of long-term sequelae.
Cytokine Release Syndrome
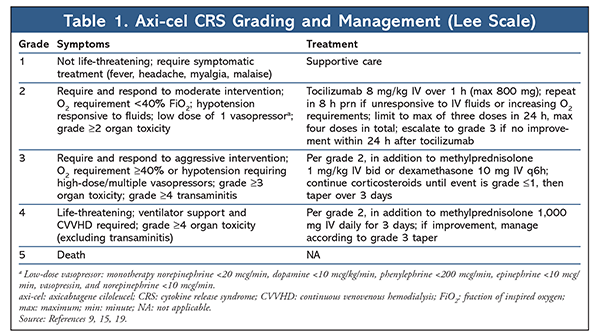
CRS has emerged as the hallmark AE of CAR-T cell therapy as a consequence of immune reconstitution from supraphysiological cytokine generation following in vivo CAR-T cell activation and proliferation. Various cytokines augment the proinflammatory cascade of CRS, and interleukin (IL)-6 has been identified as the primary mediator.5,14 The classic presentation is high fever (frequently >40°C/104°F) accompanied by constitutional or flulike symptoms. As cytokine levels continue to escalate, rapid progression to a sepsislike state with vasopressor-dependent hypotension, capillary-leak syndrome, coagulopathies, acute respiratory distress, and eventual progression to multiorgan failure may ensue. Ferritin and C-reactive protein, acute-phase reactants that are often elevated in CRS, may serve as surrogates for cytokine elevation in assessing the degree of inflammation.14-16 Symptom onset coincides with in vivo CAR-T cell proliferation. There is a direct correlation between the severity of CRS manifestations and the degree of pretreatment tumor burden.15 The Lee scale is used to grade and manage CRS severity with axi-cel, whereas clinical trials of tisagenlecleucel have used the Penn scale.15,16 Owing to classification differences between these scales, the incidence and severity of CRS reported in clinical trials of axi-cel and tisagenlecleucel cannot be directly compared. The Lee and Penn CRS grading scales are summarized in TABLES 1 and 2.

Although CRS is potentially life-threatening, its development may predict the magnitude of antitumor response. Therefore, CRS treatment requires that the physiological symptoms of uncontrolled inflammation be minimized without sacrificing CAR-T cell efficacy. Because CRS symptoms may overlap with those of sepsis and neutropenic fever, a thorough diagnostic workup including blood and urine cultures and imaging should be performed. Upon clinical suspicion of CRS, treatment and empirical antibiotics for possible infection should be initiated immediately. Fever should be managed with acetaminophen and cooling blankets. The use of nonsteroidal anti-inflammatory drugs should be avoided in patients whose platelets are below 50,000/mcL or who are at risk for kidney injury. Hypotension should be aggressively managed with fluid boluses and progression to vasopressor support as needed based on clinical response. To minimize hypotension, pharmacists can intervene by recommending that antihypertensives be held or modified prior to infusion. Although fatal bleeding events are rare, coagulopathies are common following CAR-T cell therapy, and supplementation with cryoprecipitate for hypofibrinogenemia should be provided if fibrinogen is under 100 mg/dL. Additional supportive measures may include oxygen supplementation for hypoxia or dialysis for renal failure.5,14,16
Discovery of the fundamental role IL-6 plays in CRS has made targeted treatment against this cytokine a unique innovation in CRS management. Tocilizumab, a humanized monoclonal antibody directed against IL-6 receptors that was originally approved for rheumatoid arthritis, was granted expanded FDA approval in 2017 for the management of severe or life-threatening CRS.17 Targeted inhibition of the IL-6 receptor ameliorates the systemic proinflammatory response without appearing to diminish CAR-T cell efficacy. Its demonstrated rapid and near-complete reversal of hypotension, hypoxia, and fevers within hours has rendered tocilizumab the standard of care for CRS treatment. Tocilizumab was used extensively in the clinical trials leading to FDA approval of CAR-T cell therapy. The incidence of CRS in ALL recipients receiving tisagenlecleucel was 79%, with 49% experiencing grade 3 or 4 CRS and 50% requiring at least a single dose of tocilizumab.8 The incidence of CRS with axi-cel used against DLBCL was 94%, and of these cases, 13% were grade 3 or 4 and 45% required tocilizumab.9
Corticosteroids are also effective against CRS manifestations, given their broad-spectrum anti-inflammatory and antilymphocytic characteristics. These potent immunosuppressive effects may theoretically impair the efficacy of CAR-T cell therapy and are often reserved for CRS that is unresponsive to tocilizumab.6,14,15 The dose and duration of corticosteroids that actually impair the efficacy of CAR-T cell therapy are unknown; therefore, proactive reassessment and tapering upon symptom resolution are essential in order to minimize corticosteroid exposure.
Neurologic Toxicities
CRES is a distinct neurologic complication of CAR-T cell therapy. Although the mechanism is not fully understood, it is hypothesized that increased blood-brain barrier permeability resulting from systemic inflammation enhances trafficking of lymphocytes and cytokines into the central nervous system (CNS).18 Mild manifestations include confusion, aphasia, or ataxia, with severe CRES presenting as seizures, cerebral edema, or encephalopathy. Most cases occur within 14 days after infusion, although these manifestations may be delayed up to 8 weeks and occur concurrently with or independently of CRS. Rates of any-grade CRES with axi-cel or tisagenlecleucel used against DLBCL were 87% and 39%, respectively, with 31% and 12% being grade 3 or higher.12,13
Frequent monitoring and early interventions are essential to minimizing the risk of neurologic sequelae. The diagnostic approach should center around excluding alternative etiologies for neurologic impairment, including meningitis, electrolyte disturbances, and leukemic or lymphomatous CNS involvement. Limiting the use of sedating medications (i.e., benzodiazepines, opioids) minimizes the risk of confounding neurologic assessments. Seizure is a severe manifestation that accentuates the importance of limiting exposure to medications that decrease the seizure threshold and of considering the addition of prophylactic levetiracetam.9 Despite the aforementioned risks, corticosteroids remain the mainstay of therapy. Dexamethasone is preferred for lower-grade CRES based on its excellent CNS penetration, with high-dose methylprednisolone recommended for more severe CRES.6,15 Treatment should be continued until resolution to grade <1, then tapered as tolerated. Tocilizumab may be used in cases of CRES with concurrent CRS; however, it does not appear to be effective in the management of CRES alone.
B-Cell Aplasia and Infection Risk
B-cell aplasia is an on-target, off-tumor effect caused by nonspecific depletion of normal and malignant B cells expressing CD19, which can lead to diminished antibody production (specifically hypogammaglobulinemia) and heightened infection risk.5,7 Infection risk is compounded by cumulative immunosuppression from previous chemotherapy and corticosteroid exposure. Preventive measures include antibiotic prophylaxis and immune-system reconstitution with IV gamma globulin in deficient patients.7 Prior to lymphodepleting chemotherapy, prophylaxis against Pneumocystis jirovecii with trimethoprim-sulfamethoxazole (or alternatives based on allergies/intolerances) and against herpes simplex virus with acyclovir (or alternatives) is recommended.14 During neutropenic episodes, antifungal (e.g., fluconazole or an echinocandin, if hepatotoxicity is a concern) and antibacterial prophylaxis (e.g., fluoroquinolone) may be considered. Pretreatment serologic tests for hepatitis B and C viruses are required, and HIV screening is recommended because the lentiviral vector may yield a false-positive result after CAR-T cell infusion. The use of granulocyte colony-stimulating factors has been recommended to mitigate infection risk during periods of neutropenia.14 However, by recruiting cytokine-rich myeloid cells, granulocyte-macrophage colony-stimulating factor may exacerbate CRS and therefore is avoided until CRS symptoms have resolved.8,9
Additional Toxicities
Other notable AEs associated with CAR-T cell therapy include hypersensitivity reactions and tumor lysis syndrome (TLS).14 To minimize hypersensitivity reactions, administration of acetaminophen and diphenhydramine is required 30 to 60 minutes prior to CAR-T cell infusion. Baseline assessment of TLS risk factors and initiation of prophylaxis with allopurinol and fluids should also be considered.
REMS
To safely monitor and manage serious AEs, axi-cel and tisagenlecleucel are available only through a Risk Evaluation and Mitigation Strategy (REMS) program. All members of the interprofessional healthcare team involved in the care of CAR-T cell recipients are required to undergo specialized training and certification programs on the recognition and management of CRS/CRES. To limit delays in CRS management, pharmacists should ensure that two doses of tocilizumab per patient are readily available prior to CAR-T cell infusion. Pharmacists can provide medication guides and patient education on abstaining from driving or operating heavy machinery for at least 8 weeks because of the risk of delayed CRES. Owing to the immediate attention required for CRS or CRES, the REMS requires patients to remain within 2 hours of the treatment facility for at least 4 weeks after CAR-T cell infusion. Patients must carry a wallet card at all times to ensure appropriate triage to specialized care when needed.8,9
CONCLUSION
The advent of CAR-T cell therapy has ushered in a promising new era of treatment for RR hematologic malignancies with improved response rates and durable remission in patients who historically had a poor prognosis. By virtue of its unique benefits, as well as its distinct and potentially life-threatening risks, it is critical that pharmacists be knowledgeable about the infrastructure of CAR-T cell therapy and toxicity management. As CAR-T cell therapy enters mainstream cancer immunotherapy, pharmacists can play a fundamental role in the comprehensive care of these patients.
REFERENCES
1. American Cancer Society. Key statistics for acute lymphocytic leukemia. www.cancer.org/cancer/acute-lymphocytic-leukemia/about/key-statistics.html. Accessed September 26, 2018.
2. Nguyen K, Devidas M, Cheng SC, et al. Factors influencing survival after relapse from acute lymphoblastic leukemia: a Children’s Oncology Group study. Leukemia. 2008;22(12):2142-2150.
3. National Cancer Institute. Cancer Stat Facts: Non-Hodgkin lymphoma. https://seer.cancer.gov/statfacts/html/nhl.html. Accessed September 26, 2018.
4. Crump M, Neelapu SS, Farooq U, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood. 2017;130(16):1800-1808.
5. Frey NV, Porter DL. CAR T-cells merge into the fast lane of cancer care. Am J Hematol. 2016;91(1):146-150.
6. Shank BR, Do B, Sevin A, et al. Chimeric antigen receptor T cells in hematologic malignancies. Pharmacotherapy. 2017;37(3):334-345.
7. Roberts ZJ, Better M, Bot A, et al. Axicabtagene ciloleucel, a first-in-class CAR T cell therapy for aggressive NHL. Leuk Lymphoma. 2018;59:1785-1796.
8. Kymriah (tisagenlecleucel) package insert. East Hanover, NJ: Novartis Pharmaceuticals Corp; May 2018.
9. Yescarta (axicabtagene ciloleucel) package insert. Santa Monica, CA: Kite Pharma, Inc; October 2017.
10. Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. 2015;125(26):4017-4023.
11. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439-448.
12. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531-2544.
13. Schuster SJ, Bishop MR, Tam CS, et al. Primary analysis of Juliet: a global, pivotal, phase 2 trial of CTL019 in adult patients with relapsed or refractory diffuse large B-cell lymphoma [abstract 577]. Presented at the 2017 American Society of Hematology Annual Meeting; December 11, 2017; Atlanta, GA.
14. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127(26):3321-3330.
15. Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188-195.
16. Porter D, Frey N, Wood PA, et al. Grading of cytokine release syndrome associated with the CAR T cell therapy tisagenlecleucel. J Hematol Oncol. 2018;11(1):35.
17. Actemra (tocilizumab) package insert. South San Francisco, CA: Genentech Inc; September 2018.
18. Gust J, Hay KA, Hanafi LA, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. 2017;7(12):1404-1419.
19. National Institutes of Health, National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE). Version 4.03. Bethesda, MD: National Institutes of Health; 2010. NIH Publication No. 09-5410.
To comment on this article, contact rdavidson@uspharmacist.com.
« Click here to return to Journal of Specialty Pharmacy Update.


