US Pharm. 2021;46(6):39-42.
ABSTRACT: The need to enhance the transparency of the pharmaceutical supply chain and ensure availability of generic and biosimilar drug products has become more evident during the pandemic. It is important to recognize the regulatory challenges involved in ensuring the quality and integrity of biosimilar formulations. The biosimilar market continues to grow significantly, with notable new drug approvals for 2020. An FDA spokesperson answered U.S. Pharmacist’s questions regarding the latest biosimilar regulations. Even as the FDA moves to improve access to biosimilar drug products, strategies some manufacturers use to navigate around the patent and regulatory systems to prolong the market exclusivity for brand-name products are also impacting accessibility.
The importance of enhancing the transparency of the U.S. pharmaceutical supply chain and ensuring the availability of more cost-effective generic and biosimilar drug products has only increased during the pandemic. As the medical community anticipates that more manufacturers will develop vaccines and possibly other novel biological agents to treat COVID-19, it is important to recognize the regulatory challenges involved when ensuring the quality and integrity of biosimilar formulations when compared with the innovating agent.
Generic medications have long established their role in reducing healthcare costs, not only because they are more affordable out-of-pocket for patients but also because their lower cost has improved accessibility through market competition, which stimulates further innovation and drug development. And as we begin to see the impact of vaccines and treatment medications on the COVID pandemic, it is important to reflect on the essential role these generic drugs have played during this past year and that advocacy organizations such as the Association for Accessible Medicines (AAM) have emphasized the need for a healthy generic marketplace and for policies that strengthen the sustainability of the generics and biosimilars industry.1
At this time, the energy and efforts are focused on mass vaccine distribution and administration; however, it is essential that we recognize that patients continue to receive lifesaving interventions that are comprised of generic medications used as the first-line treatments for COVID-19. These interventions include, but are not limited to, sedatives used for intubation and corticosteroids used for anti-inflammatory properties.2 To emphasize and support the importance of the generic-medicine supply chain during the COVID-19 pandemic, the AAM launched a new advocacy campaign, “Secure Our Meds,” which includes a series of reports that provide recommendations on how to get these generic and biosimilar agents to market and to sustain their availability.3 During the past year the public witnessed an unprecedented supply-chain disruption in food and basic household and personal supplies. Although not as visible to the community, this same stress and disturbance occurred within the pharmaceutical production and distribution system and prompted significant scrutiny and a recognition of the potential vulnerability to which this market could be subjected. Regardless of this perceived supply-chain susceptibility, the AAM reported that despite the increased demand and disabled shipping channels, through “innovation, perseverance, and grit, the system performed and hospitals received the drugs they needed to treat COVID-19 patients.”1
FDA IMPROVING ACCESS TO BIOSIMILARS
When discussing generic medications, it is important that the prescribing of biologics also be aligned, whenever possible, with more cost-effective versions that share the required biosimilarity. Last year’s FDA generic update in U.S. Pharmacist focused on the sunset of the decade-long grace period and ending of the Biologics Price Competition and Innovation (BPCI) Act, which was launched in 2009.4 Since March 23, 2020, the biologics approved in New Drug Applications (NDAs) that were determined to require a Biologics License Application (BLA) were removed from the Orange Book, which was previously the exclusive list of FDA-approved drug products with therapeutic equivalence. These BLA agents were added to the Purple Book, which is the list of FDA-licensed biological products that have demonstrated the necessary biosimilarity or interchangeability, as well as those agents that retained product exclusivity and thus did not have acceptable alternatives that could be prescribed.5
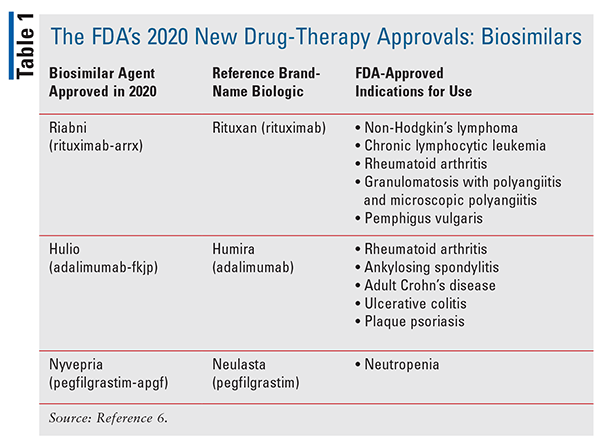
The biosimilar market continues to grow significantly, with notable new drug approvals for 2020 (TABLE 1).6 Therefore, the effort and commitment the FDA has made to increasing the information available to prescribers and patients as well as improving access to that information will hopefully encourage the consideration and prescribing of less costly alternative biological agents. Over the course of this past year, the Purple Book has been updated to include a searchable database, which was developed to ensure greater accessibility than the old table format in the previous reference.
The FDA advocates that this improved access to information provides transparency that promotes awareness about the potential alternative treatment options when there is biosimilarity or interchangeability. The transition to this more comprehensive, searchable portal has also provided links to product labeling, exclusivity details, and clinical information about dosage forms and routes of administration. Prior to the transition, the Purple Book was only available as two separate lists of the FDA-licensed biological products regulated by the Center for Drug Evaluation and Research and the FDA-licensed biological products regulated by the Center for Biologics Evaluation and Research.5
Prior to the BPCI Act, medications considered to be a biologic were submitted like any other medication regulated under section 505 of the Federal Food, Drug, and Cosmetic (FD&C) Act, however, the BPCI Act required that marketing applications for all biological products be submitted as a BLA. At the time the BPCI Act took effect, there was a 10-year transition period assigned that ended March 23, 2020.
An FDA spokesperson, Autumn Cook, answered U.S. Pharmacist’s questions regarding the latest biosimilar regulations.7
Clarify the Statutes That Give the FDA the Authority to Approve Drug and Biological-Product Medications?
There are two main statutes that provide the FDA with authority to approve drug and biological-product medications: the FD&C Act, which pertains to prescription drugs (both brand-name and generic) and OTC drugs, and the Public Health Service (PHS) Act, which applies to biological products, such as vaccines and therapeutic protein products.7
Some clarifying revisions should be included. Prior to the BPCI Act, the majority of biological-product medications were licensed under the PHS Act. However, some protein products (e.g., insulin and insulin analogs, human growth hormone, pancreatic enzymes, reproductive hormones) historically had been approved in marketing applications under the FD&C Act, which is the statutory authority under which drug products are approved. The BPCI Act required that on March 23, 2020, an approved marketing application for a biological product under the FD&C Act (which includes protein products such as insulin) be deemed to be a license for the product (i.e., an approved BLA). After March 23, 2020, all sponsors seeking approval of a biological product (that previously could have been submitted under the FD&C Act) need to submit a BLA under the PHS Act. With this transition, biological-product medications will be regulated under the same framework.
There Are Exceptions to This BLA Requirement. What Are These Exceptions, and Why Are They Necessary?
The BPCI Act describes the approval pathways that are available for submission of a marketing application for a biological product. Although there was a limited exception during a 10-year transition period ending on March 23, 2020, after that date, all sponsors seeking approval of a biological product (that previously could have been submitted under section 505 of the FD&C Act) will need to submit a marketing application under section 351 of the PHS Act: either a 351(a) BLA (i.e., a “stand-alone” BLA); or a 351(k) BLA for a proposed biosimilar product or a proposed interchangeable product.
What Is the Status of Compliance? Has There Been Any Amendment Considered to Address Challenges to Implementation?
The transition is mandated by the BPCI Act and occurred on March 23, 2020. An application holder did not need to take any affirmative steps for its approved NDA for a biological product to be deemed an approved BLA. On March 23, 2020, the FDA sent a letter to holders of approved NDAs for biological products advising that the approved NDA was deemed to be a BLA at 12:00 am Eastern daylight time on March 23, 2020, and no longer exists as an NDA.
For the past 10 years, the FDA has been working to implement the statutory transition provision in a manner that minimizes burden, helps ensure stability for patients using currently marketed products, and facilitates the development of biosimilar and interchangeable products. In addition to multiple guidances and rules issued, that can be found on the transition webpage, the FDA has also answered frequently asked questions for patients and healthcare providers. For answers to common questions about the FDA’s implementation of the “transition” provision of the BPCI Act, see the FDA Guidance for Industry, the “Deemed to be a License” Provision of the BPCI Act: Questions and Answers (March 2020). This guidance also describes the agency’s compliance policy for the labeling of biological products that are the subject of deemed BLAs, to minimize burden and possible disruption to the distribution of such products.
Why Is This Important to Pharmacists?
It is important that pharmacists understand two main things about the transition:
First, the transition was a regulatory action in which the approved drug application for a transition biological product was “deemed” to be a BLA. In general, it should not change how patients’ medications appear, or how healthcare providers prescribe or dispense their prescriptions.
Second, the biological products approved in NDAs that were deemed to be BLAs on March 23, 2020, were removed from the Orange Book (FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations) on March 23, 2020, and added to the Purple Book (FDA’s List of Licensed Biological Products with Reference Product Exclusivity and Biosimilarity or Interchangeability Evaluations). Beginning on March 23, 2020, healthcare providers can find transition biological products in the Purple Book, which identifies licensed biological products by brand name and nonproprietary name. Healthcare providers can continue using the Orange Book to identify approved drug products that are not “biological products” and related generics approved under the FD&C Act.
For more information about the transition for pharmacists, see Information for Health Care Providers About Regulatory Changes for Certain Biological Product Medications on the FDA website.
What Is the Take-Home Message for Pharmacists Regarding the FDA’s Guidance in Competitive Generic Therapies?
The FDA Reauthorization Act of 2017 (FDARA) established a new process to designate and expedite the development and review of certain drugs either intended for submission or submitted in an abbreviated new drug application (ANDA) and for which there is inadequate generic competition. It also created a new type of 180-day exclusivity for certain first-approved applicants of competitive generic therapies (CGTs). The guidance (published March 13, 2020) finalizes the FDA’s current thinking on these new statutory provisions related to CGTs first provided in the draft guidance published in February 2019.
BARRIERS TO ACCESSING BIOSIMILAR AGENTS
Even as the FDA moves to improve access to biosimilar drug products, also impacting accessibility are the strategies some manufacturers use to navigate around the patent and regulatory systems, prolonging the market exclusivity for brand-name products and impeding the FDA approval of generic alternatives and biosimilar medications.
The need for drug manufacturers to recoup their costs and to make a profit is necessary to stimulate future new-drug development and innovation. It has been reported that up to 90% of drug-development programs fail, and thus, that up to 90% of biopharmaceutical companies do not make a profit. For this reason, a blockbuster drug success can subsidize the cost of drugs that never make it to market as successful drugs. It is widely accepted that effective medicines lead to fewer physician visits, hospitalizations, surgeries, and other preventable procedures, lowering healthcare costs for society, and that new drug innovation is necessary as we live longer lives and are often faced with unexpected new health challenges such as the COVID-19 pandemic we continue to struggle through.8
Government patent protection provides the exclusive right to sell a brand-name medication for 20 years. But drug makers have found ways to extend those protections much longer. What has become a provocative and debatable contention is whether the patent holder is entitled to extended patent protection for small changes to the pre-existing approved novel agent that then leads to an ongoing market monopoly. The development of these slight variations is known as evergreening.9
The patent protection that a patent holder is entitled to, however, can be also the source of manipulation of the rules to delay generic or biosimilar agents from becoming available as a more cost-effective option. One of the most extreme examples of one of these manipulative tactics involved the main patent on the biological product Humira, which was due to expire at the end of 2016; but AbbVie, the manufacturer of Humira, developed more than 70 newer patents, a tactic known by many in the business as a “product patent thicket,” with most of these created within the few years prior to the scheduled patent expiration. These patents were reported to cover not only drug formulation but also specific disease indications and manufacturing processes.
In a recent interview in The New York Times, Richard A. Gonzalez, the company’s chief executive, stated “Any company seeking to market a biosimilar version of Humira will have to contend with this extensive patent estate, which AbbVie intends to enforce vigorously.”10 According to the AAM, product thickets make it easy for the original brand company to claim infringement and to further protect their market exclusivitiy.11 Other more flagrant stall tactics include preventing a generic company from demonstrationg interchangeability of products being compared. The refusal to provide the necessary sample of the brand-name reference biological product prevents the required demonstration of bioequivalence or biosimilarity, and blocks the company’s ability to bring the less-expensive version to market.12
CONCLUSION
The FDA has promoted their support and advocacy for the regulations that have allowed their approval of biosimilar agents to foster competition in the marketplace, expand treatment options, increase access to patients, and to lower healthcare costs overall. There have been 29 biosimilar approvals since 2015, and the number is expected to increase. For this reason, pharmacists should incorporate a periodic review of the biosimilar approvals and routinely check the Purple Book to ensure that they can advise patients of the opportunity to receive a biosimilar medication whenever possible.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
REFERENCES
1. Association for Accessible Medicines (AAM). 2021 Generics and biosimilars outlook. https://accessiblemeds.org/resources/blog/2021-generics-and-biosimilars-outlook. Accessed March 30, 2021.
2. FDA. FDA’s generic drug program in 2020 helped ensure availability of high-quality, affordable drugs amid COVID-19. FDA.gov/news-events/fda-oices/fdas-generic-drug-program=2020-helped-ensure-availability-high-quality-affordable-drugs-amid-covid#: Accessed March 30, 2021.
3. Secure our meds. www.secureourmeds.org/. Accessed March 30, 2021.
4. Demler TL. Getting up to speed with generic regulations. www.uspharmacist.com/article/getting-up-to-speed-with-generic-regulations. Accessed March 30, 2020.
5. FDA. The Purple Book. https://purplebooksearch.fda.gov/. Accessed March 30, 2021.
6. FDA. Advancing health through innovation: new drug therapy approvals 2020. Additional approvals. Biosimilars, p. 34. FDA.gov/drugs/new-drugs-fda-cder-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2020. Accessed March 30, 2021.
7. FDA. Interview with FDA spokesperson Autumn Cook, Trade Press Officer. March 1, 2021.
8. Colorado Bioscience Association. How do drug companies determine prices? https://cobioscience.com/how-do-drug-companies-determine-prices. Accessed March 9, 2021.
9. Collier R. Drug patents: the evergreening problem. CMAJ. 2013;185(9):E385-E386. Accessed March 30, 2021.
10. Makers of Humira and Enbrel using new drug patents to delay generic versions. New York Times. July 15, 2016. www.nytimes.com/2016/07/16/business/makers-of-humira-and-enbrel-using-new-drug-patents-to-delay-generic-versions.html. Accessed March 29, 2021.
11. Association for Accessible Medicines. Intellectual Property and Patent Reform. https:/accessiblemeds.org/advocacy/intellectual-property-patent-reform. Accessed May 24, 2021.
12. Federal Trade Commision. FTC files amicus brief explaining that pharmaceutical “product hopping” can be the basis for an antitrust lawsuit. www.ftc.gov/news-events/press-releases/2012/11/ftc-files-amicus-brief-explaining-pharmaceutical-product-hopping. Accessed March 30, 2021.
To comment on this article, contact rdavidson@uspharmacist.com.



