US Pharm. 2019;44(6)(Generic Drugs suppl):26-29.
ABSTRACT: Recent voluntary recalls of certain angiotensin II receptor blockers have heightened scrutiny of the non-U.S. manufacturing of drugs marketed in the U.S. and of FDA oversight of the process. Nitrosamine-based impurities that have been associated with significant health risks have been found. The FDA has determined that the presence of impurities poses an unnecessary risk to patients. It has developed new testing methods to find impurities and understand how they form, and is committed to implementing measures to prevent the formation of impurities. The FDA has been communicating the status of its various agencies’ inquiries to health professionals and the public via press releases, and responds to questions posed by U.S. Pharmacist in this article.
All drugs marketed in the United States must meet minimum quality standards that ensure every dose is effective and safe as well as free from contamination. However, recent reports about certain generic medications manufactured at approved international sites have caused heightened scrutiny of, and diminished consumer confidence in, the quality of such pharmaceuticals.1 In particular, the angiotensin II receptor blockers (ARBs) valsartan and, most recently, losartan, have been involved in voluntary recalls.
The Office of Pharmaceutical Quality (OPQ) is responsible for overseeing the quality of drugs marketed for human use in the United States. The OPQ operates within the Center for Drug Evaluation and Research (CDER) of the FDA and provides quality assurance efforts to ensure that drug products consistently meet expectations of users.1
In 2018, the FDA surveyed consumers and patients on their perception and knowledge regarding the quality and safety of drug products used in the U.S. The results of the survey revealed that roughly 75% of respondents were not confident that drugs marketed in the U.S. but manufactured outside the country had to adhere to the same strict manufacturing regulations and standards as those manufactured within the U.S. Nearly all respondents reported the belief that the imported drugs were of lesser quality than those manufactured domestically.1 Eighty percent of the FDA-registered manufacturers of active pharmaceutical ingredients (APIs) are actually located outside the U.S.1
Recalled for Impurities
The media have also played a role in communicating concerns about drug quality, with press coverage in July 2018 of the discovery of an inactive ingredient that is a suspected human carcinogen. Since then, the FDA has announced over 20 voluntary recalls of ARBs from a handful of major manufacturers including, but not limited to, Sandoz, Teva, and Mylan for nitrosamine- based impurities that have been associated with significant health risks. Among the currently recognized nitrosamine impurities are N-nitrosodimethylamine (NDMA), N-nitroso-diethylamine (NDEA), and N-nitroso-N-methyl-4-aminobutyric acid (NMBA).
NDMA: The NDMA impurity is classified as a probable human carcinogen and is an organic chemical that occurs naturally in certain foods, drinking water, air pollution, and industrial processes.2 Used as an additive in gasoline and lubricants and as an antioxidant and stabilizer in plastics, NDMA is a recognized special health hazardous substance.3
The unexpected presence of NDMA was thought to be related to changes in the way the active substance is manufactured. The FDA announced that because different manufacturing processes do not result in the production of NDMA, not all products containing the ARB valsartan were being recalled, and it encouraged consumers to contact their pharmacists and prescribers to see what action they should take.4
Unfortunately, it was not long after the initial public notice that a second notice was sent warning of another contaminant, which led to additional recalls of generic valsartan in September 2018.4 The additional impurity was identified as NDEA, another known animal and suspected human carcinogen.
NDEA: As early as 2015, 4 years prior to these official recalls, researchers reported that high doses of NDEA could cause severe injury to the liver and red blood cells in rats, which could also lead to liver damage and liver cancer. However, researchers noted that the maximum damage due to NDEA treatment was most apparent in the subjects receiving a lower dose of NDEA (100 mg/kg) for a prolonged duration (2 weeks). Therefore, it is hypothesized that duration of exposure may be more critical than the actual NDEA dose received and that hepatic damage and altered cellular functions may be the consequence of prolonged elevated oxidative stress, likely mediated by NDEA.5
NMBA: While the FDA continued to make it clear that its investigation was still ongoing, it had to issue another recall notice when yet a third impurity, NMBA, was detected. This discovery prompted Hetero Labs Ltd. of India to announce a recall of 87 lots of losartan potassium tablets, the first ARB recall associated with the newest chemical impurity.6
Exposure Thresholds
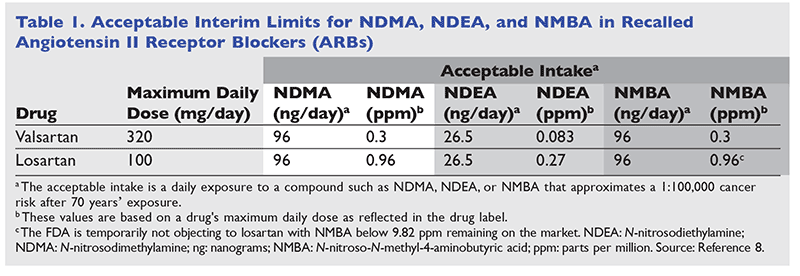
It should be noted that although the FDA has determined that the presence of these impurities poses an unnecessary risk to patients, there are still acceptable exposure thresholds of these nitrosamine impurities (TABLE 1).7 It has also been reported that nitrosamine contaminants such as NDMA are commonly found in the environment, finding their way into drinking water and food, including meats, dairy products, and vegetables. When considering the impact of the limits in medications expressed in ng/day (1,000,000 ng = 1 mg), it is also important to understand the estimated potential unintentional ingestion of these chemicals in our diet.7
Avoiding a Shortage Crisis
In order to avoid drug shortages, the FDA is allowing a temporary distribution of losartan that contains NMBA above 0.96 parts per million (ppm) but below 9.82 ppm until production of a sufficient amount of losartan can be manufactured without this impurity. Experts at the FDA are reassuring the public that this brief exposure above what was released earlier as “acceptable” poses no significant cancer risk when compared with a lifetime exposure at NMBA levels of 0.96 ppm or greater.8
The FDA is requiring manufacturers to notify the agency when lots of tested losartan exceed the interim acceptable limits in order for the FDA to determine whether these batches should be released in order to prevent further shortages. The FDA estimates that an adequate supply of nitrosamine-free losartan will be available in the U.S. in about 6 months. The FDA reports that it continues to ensure that products entering the U.S. market do not contain nitrosamine impurities by working with companies and international regulators.8 Because APIs can be manufactured outside the U.S. and then domestically incorporated into final drug products to be sold to American consumers, the FDA reports it is continually monitoring the whole process.8
FDA Communicates Concerns
The FDA has been issuing press releases describing the newest updates on the recalls due to nitrosamine impurities, and FDA Commissioner Scott Gottlieb, MD, has reassured the public that the agency continues to explore the reasons for, and potential remedies to, the presence of these potentially carcinogenic impurities.9
In a statement, Commissioner Gottlieb said, “We are deeply concerned about the presence of a third nitrosamine impurity in certain ARB medications, but it’s important to underscore that, based on the FDA’s initial evaluation, the increased risk of cancer to patients with NMBA exposure appears to be the same for NDMA exposure but less than the risk from NDEA exposure. That said, any presence of such impurities in drug products is not acceptable. Over the past few months, the FDA has conducted a major investigation and has worked with drug companies to address the presence of impurities in these products.”9
Commissioner Gottlieb further stated, “Our ongoing effort has determined that the impurities may be generated by specific chemical reactions in the manufacturing process of the drug’s active pharmaceutical ingredients. FDA scientists have developed novel and sophisticated testing methods specifically designed to detect and measure NDMA and NDEA impurities in ARB medicines. Because of the potential for discovering other nitrosamine impurities, we are conducting an extensive organic chemistry analysis to develop novel testing methods to detect additional nitrosamine impurities, including NMBA. We’re continuing to share these testing methods with international regulators, industry, and the public to help manufacturers and other regulators evaluate these products for any potential nitrosamine impurity. We are making important strides at understanding how these impurities form and we are continuing to examine if nitrosamine impurities may also arise during the manufacture of other ARB drug products. The FDA is committed to implementing measures to prevent the formation of these impurities during drug manufacturing processes in the future.”9
The FDA Responds to Questions From U.S. Pharmacist
An FDA spokesperson, Kristofer Baumgartner, provided answers to questions posed by U.S. Pharmacist.
Q: Is it expected that a generic product will be produced the same way the predecessor brand-name drug was manufactured?
A: Generic drugs are not required to use the same manufacturing process as the brand-name drug. However, as with brand-name drugs, generic-drug manufacturing facilities must meet the standards in current good manufacturing practice (CGMP) regulations. CGMPs provide for systems that assure proper design, monitoring, and control of manufacturing processes and facilities. Adherence to the CGMP regulations ensures the identity, strength, quality, and purity of drug products by requiring that manufacturers of medications adequately control manufacturing operations. The FDA reviews data in applications and conducts inspections both before and after approving drugs to ensure manufacturers comply with CGMP requirements. This helps ensure that all drug products marketed in the U.S., both brand and generic, meet the same high-quality standards, regardless of where they are manufactured. We also perform regular surveillance of both brand and generic drugs to be sure that any potential issues are readily identified and investigated.10
Q: In July 2018, the OPQ described new methods to quantify these probable human carcinogenic impurities and published these methods to provide options for companies and other regulators seeking to detect the same. Were these new methods developed just for this occasion? Who are the other regulators who would be looking for these? Is this new method something that would detect other possible impurities? Should the public be expecting other impurities, and are other assays being developed proactively?
A: Due to their chemical properties, nitrosamine impurities are difficult to detect with standard screening methods. The FDA developed and published the methods so that they can be used by manufacturers, as well as by the international regulators that we have been coordinating with on our response to this issue.10
Q: Are the manufacturing sites associated with the production of these by-product impurities all outside the U.S.?
A: Nearly all ARB API manufacturing sites are located outside the U.S., including the sites that have had APIs test positive for nitrosamine impurities.10
Q: Is the nitrosamine impurity specifically associated with just the ARB class of drugs?
A: Based on what we have learned about the manufacturing processes for ARB products where impurities have been detected, we have been reviewing manufacturing processes for other drugs to identify if they may be at risk for formation of these impurities.10
Q: The FDA recommends that patients use valsartan-containing medicines made by alternative companies if other treatment options are not available for the patient’s medical condition. Does the FDA know which process avoids the development of these by-products and can it mandate a production process?
A: The FDA’s evaluation suggests that the nitrosamines found in ARBs may be generated when specific chemicals and reaction conditions are present in the manufacturing process of the drug’s API, and may also result from the reuse of materials, such as solvents.
The FDA cannot mandate a particular production process; however, we know these drugs can be made free of nitrosamine impurities, and we are working with manufacturers to ensure they are manufacturing drugs that meet quality standards. The FDA has contacted all drug-product and API manufacturers to inform them of factors that may lead to the formation of nitrosamine impurities, and to communicate our expectation that nitrosamines should be absent—i.e., below a limit of detection—from ARBs. The interim acceptable limits we have posted for nitrosamine impurities are being used to manage recalls and avoid potential shortages.10
Q: How were these interim limits of exposure
decided upon?
A: FDA scientists reviewed publicly available safety information on each of the identified nitrosamines that have been shown to cause cancer in animals. Doses that cause cancer in animals were used to calculate an interim limit for each nitrosamine that approximates a 1:100,000 risk over a person’s lifetime. These interim limits were then adjusted based on the maximum daily dose of each ARB.10
A New Method to Detect Impurities
The FDA has updated its testing methods to detect nitrosamine impurities such as NDMA and NDEA. These new methods were initially validated for valsartan; however, the FDA has clarified that these methods can also be used for other ARB substances and drug products.11
Conclusion
The FDA promotes widespread assurance that the drugs (and APIs) manufactured outside the U.S. meet the same standards as drugs made domestically. As we continue to face ever-escalating drug costs, we are faced with the necessity to use the least expensive, yet appropriately effective, options including, but not limited to, generic drugs. It is well understood that the FDA provides oversight during premarketing, but the FDA also tests selected drug products and APIs after they are available on the market to confirm consistent quality and required potency to meet the standards established for generic drugs. In order to test for potential product failure (not meeting the quality or expected potency) for drugs manufactured outside the U.S., the FDA recently reported that agency laboratories tested 323 products from around the world, including more than 100 from India. All samples met U.S. market-quality standards using testing standards submitted in marketing applications and/or established by the United States Pharmacopeia.
REFERENCES
1. The Office of Pharmaceutical Quality (OPQ) 2018 Annual Report. www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/UCM632265.pdf. Accessed April 1, 2019.
2. Analysis of N-nitrosodimethylamine (NDMA) levels in recalled valsartan in the U.S. Press release: July 27, 2018. www.fda.gov/drugs/drugsafety/ucm613916.htm. Accessed April 1, 2019.
3. Hazardous waste sheet. https://nj.gov/health/eoh/rtkweb/documents/fs/1404.pdf. Accessed April 1, 2019.
4. FDA provides update on its ongoing investigation into valsartan products and reports on the finding of an additional impurity identified in one firm’s already recalled products. Press release: September 13, 2018. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm620499.htm. Accessed April 1, 2019.
5. Mukherjee D, Ahmad R. Dose-dependent effect of N’-Nitrosodiethylamine on hepatic architecture, RBC rheology and polypeptide repertoire in Wistar rats. Interdiscip Toxicol. 2015;8(1):1-7.
6. FDA provides update on its ongoing investigation into ARB drug products; reports on finding of a new nitrosamine impurity in certain lots of losartan and product recall. Press release: March 1, 2019. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm632425.htm. Accessed April 1, 2019.
7. FDA updates recalled valsartan-containing product information and presents NDMA levels in some foods. Press release: August 20, 2018. www.fda.gov/Drugs/DrugSafety/ucm613916.htm. Accessed April 1, 2019.
8. FDA updates table of interim limits for nitrosamine impurities in ARBs Press release: February 28, 2019. www.fda.gov/Drugs/DrugSafety/ucm613916.htm. Accessed April 1, 2019.
9. FDA updates recalled valsartan-containing and losartan-containing medicine information update. Press release: March 22, 2019. www.fda.gov/Drugs/DrugSafety/ucm613916.htm. Accessed April 1, 2019.
10. Interview with FDA spokesperson Kristofer Baumgartner, March 25, 2019.
11. FDA presents interim limits of nitrosamines in currently marketed ARBs. Press release: December 12, 2018. www.fda.gov/drugs/drugsafety/ucm613916.htm. Accessed April 1, 2019.
12. Statement from FDA Commissioner Scott Gottlieb, M.D., and Director of FDA’s Center for Drug Evaluation and Research and Janet Woodcock, M.D., on the FDA’s continuing efforts to maintain its strong oversight of generic drug quality issues domestically and abroad. Press release: February 22, 2019. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm631838.htm. Accessed April 1, 2019.
To comment on this article, contact rdavidson@uspharmacist.com.
« Click here to return to Generic Drug Update.



