US Pharm. 2020;45(6):30-32.
The FDA recently provided U.S. Pharmacist with updates on several non–COVID-19 topics, allowing a reprieve from the constant corona virus headlines that have inundated us all.1 One such topic surrounds an updated application and approval process of biological drugs manufactured and used in the United States.
On March 23, 2020, the grace period allowing full transition to compliance with the Biologics Price Competition and Innovation Act of 2009 (BPCI Act) ended. The BPCI requires that an approved marketing application for a biological product defined by the Federal Food, Drug, & Cosmetic (FD&C) Act will also be considered a license for the biological product, or an approved biologics license application (BLA), as outlined by the Public Health Service Act. The FDA brought this to our attention because the 10-year transition period should have been adequate to allow manufacturers to comply with these statutory changes, thus ensuring biological products submitted are approved for use and meet all requirements of section 505 of the FD&C Act.2,3 (Prior to the BPCI Act, medications considered to be biologics were submitted like any other medication under section 505 of the FD&C Act.)
The FDA defines a drug as a substance that demonstrates efficacy in the treatment, prevention, or cure for a disease.4 The new wave of biological drugs is consistent with this definition. The differentiating factor is that unlike traditional medications, which are well-defined, chemically synthesized agents, biological drugs are developed using originating substances that are much less well defined. Often derived from “living material consisting of microorganisms, human or animal tissues,” these agents are described as a “virus, therapeutic serum, toxin, antitoxin, vaccine, blood, blood component or derivative, allergenic product, or analogous product applicable to the prevention, treatment, or cure of a disease or condition of human beings.”5
In March, the FDA Published Guidance for Competitive Generic Therapies. What Is the Take-Home Message?
According to FDA representative Charles Kohler, the FDA Reauthorization Act of 2017 established a new process to designate and expedite the development and review of certain drugs either intended for submission or submitted in an abbreviated new drug application (ANDA) and for which there is inadequate generic competition.1 It also created a 180-day exclusivity for certain first-approved applicants of competitive generic therapies (CGTs). The guidance finalizes the FDA’s current thinking on these new statutory provisions related to CGTs, first provided in the draft guidance published in February 2019.
Are Biosimilar Agents Generic Biological Products?
According to the FDA website, biosimilars are biological products that are considered highly similar to one another. This similarity equates to no “clinically meaningful differences” when compared with the current reference product, which is the single-source biological product already approved by the FDA. When first approved, the reference product must demonstrate adequate effectiveness, as well as safety. There are many unique challenges encountered when comparing and evaluating these similar agents, including, but not limited to, what is considered to be “within-product” variations that are viewed as expected variations based not only on the behavior of the biological product being developed but also those resulting from the manufacturing process, which can have more variables when compared with the production of traditional “smaller molecule” drugs.5
According to the FDA, roughly 90% of prescriptions filled in the U.S. are generic, which translates into greater access to medication and more affordable care. The FDA has promoted efforts to increase the availability of generic drugs as an opportunity to both create competition in the marketplace and increase access to more affordable healthcare.
What Does It Mean to Be “Highly Similar”?
According to the FDA, minor differences in what would be considered “clinically inactive” ingredients between the biosimilar and reference product are acceptable. Examples offered by the FDA of these inactive (inert) differences include buffers or stabilizers. The FDA monitors, reviews, and controls both the “within-product” variations, which occur during manufacturing for both reference and biological products, and also “within-product” differences, such as those seen in different lot batches of the same drug.6
What Are “Clinically Meaningful Differences”?
According to the FDA, “clinically meaningful differences” include evidence that the proposed biosimilar agent exhibits significant departures of potency, compromising effectiveness and/or safety, as well as purity. These differences generally arise from response and exposure studies examining pharmacokinetics and pharmacodynamics of the medications under review.6
What Is an Interchangeable Product?
According to the FDA, for a biosimilar agent to claim interchangeability, it must meet additional requirements outlined by the BPCI Act, with some of these elements including, but not limited to, demonstrating the same clinical outcomes (medical “results”) as the original reference product. The FDA also considers ongoing safety and efficacy of longitudinal interchange, an example of which would be a patient alternating use of the different products over time. While state laws regarding generic substitution differ, some allow patients who prefer a less expensive biosimilar product that is approved by FDA as interchangeable to request their pharmacist to substitute the interchangeable product for the reference product without contacting the prescriber.7
New Treatment Indications for Approved Drugs
With the recent wave of interest examining the potential new role of older drugs, such as chloroquine phosphate and hydroxychloroquine for the possible treatment and/or prevention of COVID-19, questions have arisen regarding what the FDA will require to transition this older generic drug into a new treatment in the future. To answer thess questions, we need to understand the process by which the FDA approaches the approval of a generic drug in general and for one that is being repurposed. In addition, it is helpful to consider the current thinking of the FDA with regard to such repurposing and the priority the FDA gives to the generic-drug approval process overall.
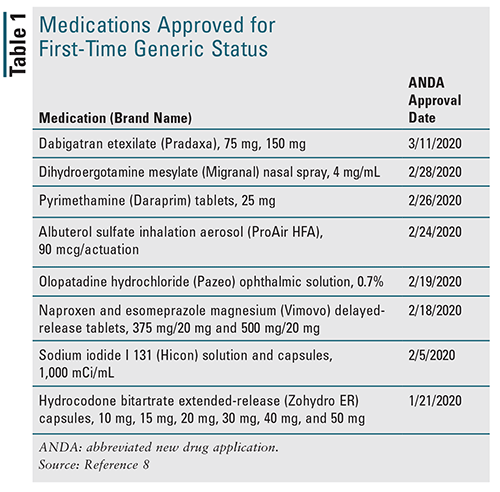
The FDA’s Center for Drug Evaluation and Research (CDER) is responsible for approving medication applications and prioritizing the review of the submissions for first generic drug approvals. In the first quarter of 2020 alone, there have been eight generic approvals for drugs previously exclusively available as brand-name medications (TABLE 1).8 In a pre–COVID-19 pandemic interview with The ASCO Post, in partnership with the American Society of Clinical Oncology (ASCO), then FDA Commissioner Scott Gottleib, MD, communicated the need for evidence-based, multidisciplinary cancer care and opportunities to apply new ways of thinking about drug development and regulations surrounding the process.9
REFERENCES
1. FDA. Surgical mask and gown conservation strategies—letter to healthcare providers. www.fda.gov/medical-devices/letters-health-care-providers/surgical-mask-and-gown-conservation-strategies-letter-healthcare-providers. Accessed March 28, 2020.
2. FDA. Deemed to be a license provision of the BPCI Act. www.fda.gov/drugs/guidance-compliance-regulatory-information/deemed-be-license-provision-bpci-act. Accessed March 28, 2020.
3. Biological Products Regulated Under Section 351 of the Public Health Service Act; Implementation of Biologics License; Elimination of Establishment License and Product License; Technical Amendment. www.federalregister.gov/documents/2000/08/29/00-21895/biological-products-regulated-under-section-351-of-the-public-health-service-act-implementation-of-.
4. FDA. Generic drugs. www.fda.gov/drugs/buying-using-medicine-safely/generic-drugs. Accessed March 13, 2020.
5. The Federal Register. www.federalregister.gov/reader-aids/using-federalregister-gov. Accessed March 28, 2020.
6. FDA. Frequently asked questions about therapeutic biological products. www.fda.gov/drugs/therapeutic-biologics-applications-bla/frequently-asked-questions-about-therapeutic-biological-products. Accessed March 28, 2020.
7. FDA. What is a biological product? www.fda.gov/drugs/biosimilars/biosimilar-and-interchangeable-products#biological.
8. FDA. First generic drug approvals. www.fda.gov/drugs/drug-and-biologic-approval-and-ind-activity-reports/first-generic-drug-approvals. Accessed April 1, 2020.
9. FDA helps streamline approval process for supplemental drug indications. https://ascopost.com/issues/december-25-2017/fda-helps-streamline-approval-process-for-supplemental-drug-indications. Accessed April 1, 2020.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
To comment on this article, contact rdavidson@uspharmacist.com.



