US Pharm. 2019;44(10):18-28.
New molecular entities (NMEs), as defined by the FDA, are new drug products containing as their active ingredient a chemical substance marketed for the first time in the United States. The following descriptions of NMEs approved in 2018–2019 (TABLE 1) details the basic clinical and pharmacologic profiles of each new drug, as well as key precautions and warnings. Also included is a brief summary of selected pharmacokinetic, adverse-reaction, drug-interaction, and dosing data submitted to the FDA in support of the manufacturer’s New Drug Application. This review is intended to be objective rather than evaluative in content. The information for each NME was obtained primarily from sources published prior to FDA approval. Experience clearly demonstrates that many aspects of a new drug’s therapeutic profile are not detected in premarketing studies and emerge after the drug is used in large numbers of patients. Studies have demonstrated the appearance of “new” adverse reactions for many NMEs within several years after they first become available. Some of these drugs may eventually acquire at least one black box warning for serious adverse drug reactions or are withdrawn from the market for safety reasons that were not recognized at the time of approval. Therefore, while this review offers a starting point for learning about new drugs, it is essential that practitioners be aware of changes in a drug’s therapeutic profile as reported in the pharmaceutical literature and by their own patients.
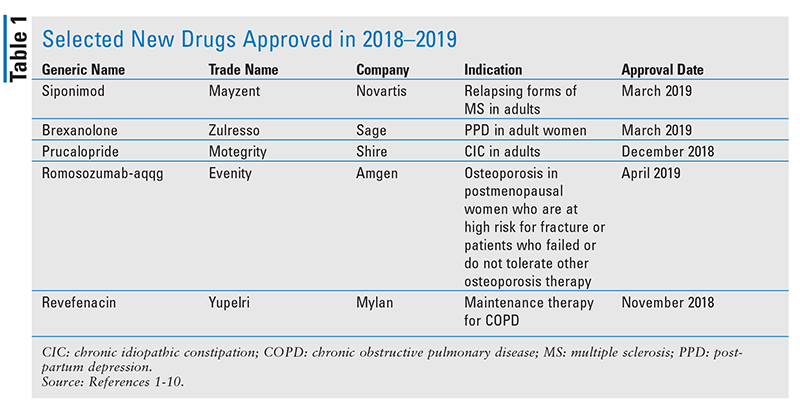
Siponimod (Mayzent, Novartis)
Indication and Clinical Profile1,2: Siponimod is indicated for the treatment of relapsing forms of multiple sclerosis (MS) in adults, including clinically isolated syndrome, relapsing-remitting MS (RRMS), and active secondary progressive MS (SPMS). MS is a chronic autoimmune disease of the central nervous system in which the immune system destroys the myelin sheath that surrounds the nerve cells in the brain and spinal cord. This demyelination slows or even stops nerve impulses, resulting in muscular weakness or motor-skill dysfunction, vision problems, and cognitive dysfunction. It is estimated that 1 million Americans have MS, and most cases (85%) begin as RRMS, in which episodes of worsening function are followed by recovery periods in which some residual disability may remain. Over time, up to 90% of patients with RRMS will transition to SPMS, which involves worsening neurologic function independent of relapses during the disease course. In the initial stage of SPMS, patients may continue to experience relapses (active SMPS) for several years, after which relapses no longer occur but disability progresses (inactive SMPS).
FDA approval was based on data from a randomized, double-blind, parallel-group, placebo-controlled trial involving 1,651 subjects with SPMS who had evidence of disability progression in the prior 2 years, no relapses 3 months before enrollment, and an Expanded Disability Status Scale (EDSS) score between 3.0 and 6.5. The primary endpoint was time to 3-month confirmed disability progression (CDP), defined as a ≥1-point increase in EDSS from baseline (0.5-point increase for patients with baseline EDSS of ≥5.5) sustained for 3 months. Evaluations were performed at screening, every 3 months during the study, and when a suspected relapse occurred. MRI evaluations were performed at screening and every 12 months. At study completion, based on a time-to-event analysis, 6% fewer patients (statistically significant) experienced 3-month CDP compared with placebo. Siponimod patients also experienced a reduction in annualized relapse rate compared with placebo (0.071 vs. 0.160, respectively). The safety and efficacy of siponimod have not been studied in pediatric patients.
Pharmacology and Pharmacokinetics1,2: Siponimod (FIGURE 1) is an azetidinecarboxylic acid that acts as a high-affinity sphingosine-1-phosphate (S1P) receptor 1 and 5 modulator. Its action at these lymph node receptors prevents the egress of lymphocytes into peripheral circulation and, possibly, migration into the central nervous system.
Siponimod has an absolute oral bioavailability of 84%, and it achieves peak plasma concentration about 4 hours post dose. It is extensively metabolized—primarily via CYP2C9 (79%) and 3A4 (19%)—to inactive metabolites with subsequent biliary or fecal excretion. Siponimod is cleared at a mean rate of 3.11 L per hour and has an elimination half-life of roughly 30 hours.
Adverse Reactions and Drug Interactions1,2: The most common (≥10%) adverse reactions in clinical trials were headache, hypertension, falls, and serum transaminase elevation. Other potential serious adverse reactions included increased risk of infection, possibly irreversible reductions of forced expiratory volume in 1 second, elevations of liver enzymes, and macular edema. Siponimod may cause cardiovascular adverse effects including bradycardia, atrioventricular (AV) conduction delay, and hypertension (which are typically mild and resolve during treatment), as well as QT prolongation. Because of these effects, a cardiology consultation is recommended before siponimod is initiated in patients with a QT interval >500 msec, arrhythmias requiring treatment with Class Ia or III antiarrhythmic drugs, ischemic heart disease, heart failure (HF), history of cardiac arrest or myocardial infarction (MI), cerebrovascular disease, uncontrolled hypertension, history of second-degree or higher AV block, sick sinus syndrome, or sinoatrial heart block. The use of siponimod with other medications that prolong the QT interval or are known to decrease heart rate (e.g., verapamil, diltiazem, beta-blockers) should be avoided in the absence of a cardiology consultation owing to the effects of siponimod on heart rate.
Because of its immunosuppressive effects, siponimod should be used with caution in patients receiving antineoplastic, immune-modulating, or immunosuppressive therapy, and it should not be used following treatment with alemtuzumab. Live attenuated vaccines should be avoided during and up to 4 weeks after treatment with siponimod, and treatment should be paused at least 1 week before and for 4 weeks after administration of other vaccines, as siponimod may reduce their effectiveness. Given its mechanism of action, patients receiving siponimod should be monitored for posterior reversible encephalopathy syndrome, progressive multifocal leukoencephalopathy due to the John Cunningham virus, and rebound syndrome, as the use of other S1P receptor modulators has resulted in these adverse effects.
Because of its extensive CYP metabolism, siponimod exposure can be altered by drugs that affect these enzymes; therefore, siponimod should not be used concomitantly with drugs that cause a combination of moderate CYP2C9 inhibition and moderate-to-strong CYP3A4 inhibition (e.g., fluconazole) or a combination of moderate CYP2C9 induction and strong CYP3A4 induction (e.g., carbamazepine). Siponimod should be used with caution with moderate CYP2C9 inhibitors or inducers.
Dosage and Administration1,2: Siponimod is supplied as 0.25-mg and 2-mg tablets for oral administration. Owing to its high level of CYP2C9 metabolism, patients should be tested for CYP2C9 variants to determine the CYP2C9 genotype. The recommended initial dosage of siponimod is 0.25 mg daily, with incremental increases up to a maintenance dosage of 1 or 2 mg daily, depending on CYP2C9 genotype. Siponimod is contraindicated in patients with a CYP2C9 *3/*3 genotype; those with a recent diagnosis of MI or unstable angina, stroke, transient ischemic attack, decompensated HF requiring hospitalization, or Class III or IV HF; and those with Mobitz type II second-degree, third-degree AV block, or sick sinus syndrome (unless patient has a functioning pacemaker). No dose adjustments are required for renal or hepatic impairment.
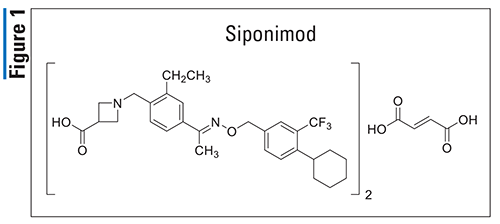
Brexanolone (Zulresso, Sage)
Indication and Clinical Profile3,4: Brexanolone, which is indicated for the treatment of postpartum depression (PPD) in adult women, is the first drug the FDA has approved specifically for this disorder. PPD is a major depressive episode (MDE) that occurs following childbirth, although its symptoms can start during pregnancy. As with other forms of depression, PPD is characterized by sadness and anhedonia (loss of interest in activities and decreased ability to feel pleasure); it typically presents with symptoms such as cognitive impairment, feelings of worthlessness or guilt, or suicidal ideation.
The efficacy of brexanolone was demonstrated in two multicenter, placebo-controlled studies in women aged 18 to 45 years who had PPD. Patients met Diagnostic and Statistical Manual of Mental Disorders criteria for an MDE with onset of symptoms in the third trimester or within 4 weeks of delivery. In both studies, patients received a 60-hour continuous IV infusion of brexanolone or placebo and were assessed for the next 4 weeks. The first study included patients with severe PPD (Hamilton Depression Rating Scale [HAM-D] score of ≥26); the second study included patients with moderate PPD (HAM-D score of 20-25). Titration to the recommended target dosage of 90 mcg/kg/h was evaluated in both studies (patients received 30 mcg/kg/h for 4 h, 60 mcg/kg/h for 20 h, 90 mcg/kg/h for 28 h, a taper to 60 mcg/kg/h for 4 h, and then 30 mcg/kg/h for 4 h). The first study also evaluated titration to a target dosage of 60 mcg/kg/h (patients received 30 mcg/kg/h for 4 h, 60 mcg/kg/h for 52 h, and then 30 mcg/kg/h for 4 h). The primary endpoint was the mean change from baseline in depressive symptoms as measured by the HAM-D total score at the end of the infusion (hour 60). In both studies, titration to a target dosage of brexanolone 90 mcg/kg/h was superior to placebo in improvement of depressive symptoms. This was also observed at the end of the 30-day follow-up period.
Pharmacology and Pharmacokinetics3,4: Brexanolone (FIGURE 2) is a progesterone derivative that appears to function as a neurohormone. This agent’s mechanism of action in the treatment of PPD is not fully understood but is thought to be related to its positive allosteric modulation of both synaptic and extrasynaptic gamma-aminobutyric acid (GABA)A receptors. Allosteric modulation of these receptors results in varying degrees of desired activity rather than complete receptor activation or inhibition. Brexanolone seems to have little or no action at other ligand-gated ion channels, including N-methyl-D-aspartate, AMPA, kainite, and glycine receptors.
Brexanolone exhibits dose-proportional pharmacokinetics over a range of 30 mcg/kg/h to 270 mcg/kg/h (3 × maximum recommended dosage). Mean steady-state exposure at 60 mcg/kg/h and 90 mcg/kg/h was about 52 ng/mL and 79 ng/mL, respectively. The drug has a volume of distribution of 3 L/kg and is >99% bound by plasma proteins. Brexanolone is extensively metabolized by non–CYP-based pathways via ketoreduction (aldo-keto reductases), glucuronidation (uridine diphosphate glucuronosyltransferases), and sulfation (cytosolic sulfotransferases), and the metabolites are pharmacologically inactive. The drug has an elimination half-life of approximately 9 hours and a total plasma clearance of 1 L/h/kg. It is excreted primarily as metabolites in the feces (47%) and urine (42%).
Adverse Reactions and Drug Interactions3,4: The most common adverse reactions (>5%) reported by patients treated with brexanolone in clinical trials included sedation/somnolence, dry mouth, loss of consciousness, and flushing/hot flush. The drug label has a black box warning stating that patients are at risk for excessive sedation or sudden loss of consciousness during administration of brexanolone. Because of these risks, patients should be monitored for excessive sedation and sudden loss of consciousness and have continuous pulse oximetry monitoring. Brexanolone is a Schedule IV controlled substance that has been shown to have an abuse-related neuropharmacologic profile similar to that of alprazolam and midazolam. Data are not available regarding use in pregnant women; however, based on findings in animals of other drugs that enhance GABAergic inhibition, brexanolone may cause fetal harm. Data from a lactation study in 12 women indicate that brexanolone is transferred to breast milk in nursing mothers; however, the relative infant exposure is low (1%-2% of the maternal weight-adjusted dosage). Available data do not suggest a significant risk of adverse reactions from exposure in breastfed infants.
Coadministration of brexanolone with central nervous system depressants (opioids, benzodiazepines) may increase the likelihood or severity of sedation. In the placebo-controlled studies, a higher percentage of brexanolone-treated patients who used concomitant antidepressants reported sedation-related events. No clinically significant differences in the pharmacokinetics of phenytoin (CYP2C9 substrate) were observed when it was used concomitantly with brexanolone. No studies were conducted to evaluate the effects of other drugs on brexanolone pharmacokinetics.
Dosage and Administration3,4: Brexanolone is supplied as a solution for IV administration and is available only through the Zulresso Risk Evaluation and Mitigation Strategy (REMS), a restricted program that requires the drug to be administered in a certified healthcare facility registered in the program. Brexanolone is administered as a continuous IV infusion over 60 hours (2.5 days). The manufacturer’s prescribing information should be consulted for details on drug administration. As noted previously, because of the risk of serious harm due to the sudden loss of consciousness, the patient must be monitored for excessive sedation and sudden loss of consciousness and have continuous pulse oximetry monitoring. Treatment should be initiated early enough in the day to allow for recognition of excessive sedation. If excessive sedation occurs at any time, the infusion should be stopped until symptoms resolve. The infusion may be resumed at the same or lower dose as clinically appropriate. If pulse oximetry reveals hypoxia, the infusion should be stopped and not resumed. Brexanolone should be discontinued in patients whose PPD worsens or who experience emergent suicidal thoughts and behaviors. No dose adjustment is necessary in mild-to-severe renal impairment (estimated glomerular filtration rate [eGFR] ≥15 mL/min/1.73 m²), but use in end-stage renal disease (eGFR <15 mL/min/1.73 m²) should be avoided owing to potential accumulation of the solubilizing agent used for brexanolone (i.e., betadex sulfobutyl ether sodium). No dose adjustment is necessary in hepatic impairment. During the infusion periods, the patient must be accompanied while caring for her child(ren). The need for these steps is addressed in a boxed warning in the drug’s prescribing information. As part of the REMS, patients should be counseled on the risks of brexanolone treatment and instructed that they must be monitored at a healthcare facility for the entire 60 hours of infusion. After treatment, the patient should not drive, operate machinery, or do other dangerous activities until feelings of sleepiness from the treatment have completely resolved.
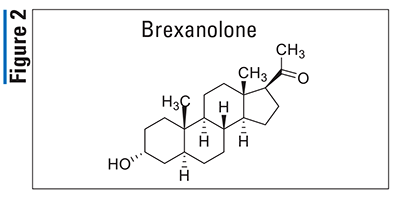
Prucalopride (Motegrity, Shire)
Indication and Clinical Profile5,6: Prucalopride is indicated for the treatment of chronic idiopathic constipation (CIC) in adults. CIC, also known as functional constipation, is characterized by difficult, infrequent, or incomplete defecation that lacks an apparent physical or physiological cause. It is estimated that approximately 35 million U.S. adults have CIC. Prucalopride is the fourth prescription drug to receive FDA approval specifically for treatment of CIC, and it is the first agent in its pharmacologic class to be approved for this use.
FDA approval was based on data from six multicenter, randomized, double-blind, placebo-controlled trials involving 2,484 adult patients with a history of CIC. The primary endpoint of all trials was the proportion of patients receiving prucalopride versus placebo who were treatment responders (i.e., experienced an average of at least three complete spontaneous bowel movements [CSBMs] per week) over 12 weeks. A statistically significant increase in the proportion of patients who responded to treatment with prucalopride (19%-38%) versus placebo (10%-18%) was demonstrated in five of the trials; the sixth trial, showing a 5% difference in responders, did not achieve statistical significance. Improvement in the frequency of CSBMs per week was seen as early as week 1 and was maintained through week 12 in all trials.
Pharmacology and Pharmacokinetics5,6: Prucalopride (FIGURE 3), a dihydrobenzofuran-7-carboxamide, is a selective serotonin-4 receptor agonist that activates lower-gastrointestinal (GI)-tract smooth muscle, resulting in stimulation of colonic peristalsis and increased bowel motility. Prucalopride has an absolute oral bioavailability of 90% and achieves peak plasma concentrations within 2 to 3 hours following administration. It undergoes minimal metabolism to at least seven different minor metabolites via CYP3A4, and 60% to 65% of the dose is excreted unchanged. Prucalopride has a half-life of 24 hours and it is primarily excreted renally (84%) via passive filtration and active secretion, with roughly 13% excreted in the feces.
Adverse Reactions and Drug Interactions5,6: The most common (≥2%) adverse reactions reported in clinical trials were headache, abdominal pain, nausea, diarrhea, abdominal distention, dizziness, vomiting, flatulence, and fatigue. Patients who experience diarrhea that is severe or persists for several days should discontinue prucalopride and consult their healthcare provider. Other adverse reactions noted during clinical trials included abnormal GI sounds, decreased appetite, migraine, and pollakiuria. Because suicidal ideation and suicide attempts were reported during clinical trials, patients who are receiving prucalopride should be monitored for worsening depression or emergence of suicidal thoughts and behaviors. Owing to its limited CYP metabolism and highly selective receptor activity, prucalopride has no known significant drug interactions.
Dosage and Administration5,6: Prucalopride is supplied as 1-mg and 2-mg oral tablets. The recommended dosage is 2 mg for patients with a creatinine clearance (CrCl) of ≥30 mL/minute and 1 mg daily for those with CrCl <30 mL/minute who do not require hemodialysis. Prucalopride use should be avoided in patients with end-stage renal disease requiring hemodialysis. The safety and efficacy of prucalopride have not been studied in pediatric patients. The drug is contraindicated in patients with a previous serious hypersensitivity reaction to prucalopride or to any components of the drug, and in those with intestinal perforation or obstruction due to structural or functional disorder of the gut wall, obstructive ileus, or severe inflammatory conditions of the GI tract.
Romosozumab-aqqg (Evenity, Amgen)
Indication and Clinical Profile7,8: The FDA approved romosozumab-aqqg for the treatment of osteoporosis in postmenopausal women who are at high risk for fracture (i.e., history of osteoporotic fracture or multiple risk factors for fracture) and in patients who failed or are intolerant to other available osteoporosis therapy. More than 10 million people in the United States have osteoporosis, which occurs most frequently in women who are past menopause.
FDA approval of romosozumab-aqqg was based on two phase III, placebo-controlled trials: FRAME and ARCH. FRAME (fracture study in postmenopausal women with osteoporosis) evaluated the efficacy of romosozumab-aqqg (210 mg administered monthly) in reducing the incidence of new vertebral fractures through 12 months in 7,180 postmenopausal women with osteoporosis. It also investigated the efficacy of romosozumab-aqqg treatment for 12 months followed by denosumab for 12 months in reducing the incidence of new vertebral fractures through 24 months. In this trial, 1 year of romosozumab-aqqg treatment lowered the risk of a new vertebral fracture by 73% compared with placebo. This benefit was maintained over the second year of the trial, when romosozumab-aqqg was followed by 1 year of denosumab therapy compared with placebo followed by denosumab.
ARCH (active-controlled fracture study in postmenopausal women with osteoporosis at high risk of fracture) was an alendronate-controlled trial of romosozumab-aqqg in 4,093 postmenopausal women with osteoporosis and previous fracture history. This event-driven trial compared 12 months of romosozumab-aqqg (210 mg monthly) followed by ≥12 months of alendronate (70 mg) with alendronate alone to assess the drug’s efficacy in reducing risk of clinical fracture (nonvertebral fracture and symptomatic vertebral fracture) through the primary analysis period and the incidence of new vertebral fracture at 24 months. In ARCH, 1 year of romosozumab-aqqg followed by 1 year of alendronate reduced the risk of a new vertebral fracture by 50% versus 2 years of alendronate alone. Romosozumab-aqqg followed by alendronate also reduced the risk of nonvertebral fractures versus alendronate alone and significantly increased bone mass density.
Pharmacology and Pharmacokinetics7,8: Romosozumab-aqqg is a humanized monoclonal antibody (immunoglobulin G2) that inhibits sclerostin, a glycoprotein secreted by osteocytes that produces antianabolic effects on bone. This drug increases bone formation and, to a lesser extent, decreases bone resorption. In animal studies, romosozumab-aqqg increased new trabecular and cortical bone formation by stimulating osteoblastic activity, resulting in increased bone mass and improvements in bone structure and strength. The bone-forming effect of romosozumab-aqqg wanes after 12 doses, so no more than 12 doses should be given. If osteoporosis therapy is needed after the 12th dose, the patient should begin a treatment that reduces bone resorption.
Peak plasma concentration (22.2 mcg/mL) occurs 5 days after SC administration of standard doses of romosozumab-aqqg, and steady state is achieved by 3 months. Development of antibodies to romosozumab-aqqg leads to decreased mean concentrations up to 22%. The drug’s volume of distribution is 3.9 L. While the metabolism has not been characterized, romosozumab-aqqg is expected to be degraded into small peptides and amino acids via catabolic pathways similar to those for endogenous IgG. The drug’s elimination half-life is 12.8 days, and its clearance is 0.38 mL/h/kg.
Adverse Reactions and Drug Interactions7,8: Common side effects reported for romosozumab-aqqg included joint pain, headache, and injection-site reactions. The drug was associated with an increased risk of cardiovascular (CV) death, heart attack, and stroke in the alendronate trial, but not in the placebo trial. Therefore, romosozumab-aqqg has a boxed warning that it may increase the risk of heart attack, stroke, and CV death and should not be used in patients who had a heart attack or stroke within the previous year. In patients with other CV risk factors, potential benefits versus risk should be assessed. If a patient experiences a myocardial infarction or stroke during therapy, romosozumab-aqqg should be discontinued. In rodent reproduction studies, weekly administration during the period of organogenesis at exposures exceeding 32 times the typical clinical exposure produced skeletal abnormalities in offspring. There is no information on the presence of romosozumab-aqqg in human milk, the effects on breastfed infants, or the effects on milk production. Therefore, this drug is not indicated for use in women of reproductive potential or those who are nursing.
Dosage and Administration7,8: Romosozumab-aqqg is supplied as 105 mg/1.17 mL single-use, prefilled syringes for SC administration and should be administered by a healthcare provider. The recommended dosage is 210 mg administered SC in the abdomen, thigh, or upper arm. Two separate prefilled syringes (each containing 105 mg of drug in 1.17 mL) are provided, and they should be injected in succession to meet the total dosage of 210 mg. The drug should be administered once per month for 12 consecutive months. Patients should be adequately supplemented with calcium and vitamin D during treatment, and preexisting hypocalcemia should be corrected before therapy is initiated. If a dose is missed, it should be administered as soon as it can be rescheduled. Thereafter, romosozumab-aqqg may be scheduled every month from the date of the last dose. No dose adjustment is required; however, patients with severe renal impairment (estimated glomerular filtration rate 15-29 mL/min/1.73 m²) or who are receiving dialysis have a greater risk of hypocalcemia.
Revefenacin (Yupelri, Mylan)
Indication and Clinical Profile9,10: Revefenacin is indicated for maintenance treatment of patients with chronic obstructive pulmonary disease (COPD). This lung disease is characterized by progressively worsening airflow obstruction that interferes with breathing. In COPD, long-term exposure to respiratory irritants, most commonly cigarette smoke, damages the lungs and airways and causes chronic bronchitis and/or emphysema. Most patients who develop COPD are aged 40 years or older, and patients typically experience chest tightness, chronic cough, and excessive phlegm. According to the CDC, 16 million Americans have COPD. According to the Heart, Lung, and Blood Institute, COPD is the fourth leading cause of death in the United States.
FDA approval was based primarily on data from two replicate 12-week, randomized, double-blind, parallel-group, placebo-controlled trials involving 1,229 subjects aged 40 years or older with moderate-to-very-severe COPD, a history of smoking for ≥10 pack-years, and a forced expiratory volume in 1 second (FEV1)/forced vital capacity ratio of ≤0.7. The primary endpoint of both trials was the change from baseline in predose FEV1 at day 85. Upon completion of the trial, revefenacin patients experienced a mean change in predose FEV1 from baseline of +127, which demonstrated a statistically significant improvement over patients who received placebo (–19). The safety and efficacy of revefenacin have not been studied in pediatric patients.
Pharmacology and Pharmacokinetics9,10: Revefenacin (FIGURE 4) is a structurally novel, nonselective, long-acting, muscarinic antagonist that blocks M3 receptors at the smooth muscle in the airways. Antagonism of these receptors blocks endogenous acetylcholine-induced bronchoconstriction, leading to net bronchodilation.
The time to peak plasma concentration of revefenacin and its active metabolite occurs 14 to 41 minutes after inhaled administration. Revefenacin and its active metabolite are extensively distributed to tissues and are 71% and 42%, respectively, plasma protein bound. The drug is primarily metabolized hepatically via hydrolysis to its major active metabolite, which maintains 10% to 33% of revefenacin’s receptor activity. Revefenacin has a terminal half-life of 22 to 70 hours, and it is excreted predominantly in the feces, with less than 1% excreted renally.
Adverse Reactions and Drug Interactions9,10: The most common (1%-10%) adverse reactions reported in clinical trials were headache, nasopharyngitis, upper respiratory tract infection, back pain, bronchitis, oropharyngeal pain, dizziness, and hypertension. Patients receiving revefenacin who experience paradoxical bronchospasm should discontinue the drug and initiate therapy with another agent. Revefenacin may cause an increase in intraocular pressure; therefore, it should be used with caution in patients with open-angle glaucoma. Owing to its potential for causing or worsening urinary retention, revefenacin should be used with caution in patients with preexisting benign prostatic hyperplasia or bladder-neck obstruction.
Coadministration of revefenacin with other anticholinergics should be avoided because of the potential for increased anticholinergic adverse effects of both medications. Concomitant use of revefenacin and drugs that inhibit organic anion–transporting polypeptide 1B1 and 1B3 (e.g., rifampicin, cyclosporine) is not recommended, as these drugs could result in increased systemic exposure of the active metabolite and in adverse effects.
Dosage and Administration9,10: Revefenacin is supplied as 175 mcg/3 mL unit-dose vials of nebulization solution. The recommended dosage is 175 mcg (1 vial) per day via a standard jet nebulizer with a mouthpiece connected to an air compressor. The use of revefenacin in patients with hepatic impairment is not recommended, and the drug is contraindicated in patients with a previous serious hypersensitivity reaction to revefenacin or to any of its components.
REFERENCES
1. Mayzent (siponimod) package insert. East Hanover, NJ: Novartis Pharmaceuticals Corp; March 2019.
2. Kappos L, Bar-Or A, Cree BAC, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet. 2018;391:1263-1273.
3. Zulresso (brexanolone) package insert. Cambridge, MA: Sage Therapeutics, Inc; March 2019.
4. Meltzer-Brody S, Colquhoun H, Riesenberg R, et al. Brexanolone injection in post-partum depression: two multicentre, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet. 2018;392:1058-1070.
5. Motegrity (prucalopride) package insert. Lexington, MA: Shire US, Inc; December 2018.
6. Mahajan R. Prucalopride: a recently approved drug by the Food and Drug Administration for chronic idiopathic constipation. Int J Appl Basic Med Res. 2019;9:1-2.
7. Evenity (romosozumab-aqqg) package insert. Thousand Oaks, CA: Amgen Inc; April 2019.
8. Cosman F, Crittenden DB, Adachi JD, et al. Romosozumab treatment in postmenopausal women with osteoporosis. N Engl J Med. 2016;375(16):1532-1543.
9. Yupelri (revefenacin) package insert. Morgantown, WV: Mylan Specialty LP; November 2018.
10. Pudi K, Pendyala S, Barnes C, et al. Trials in progress: two 12-week, randomized, double-blind, placebo-controlled, parallel-group phase 3 trials of a nebulized long-acting muscarinic antagonist (revefenacin) in study participants with moderate to very severe COPD. Chest. 2016;150:825A [abstract].
To comment on this article, contact rdavidson@uspharmacist.com.



