US Pharm. 2019;44(8):HS-1-HS-10.
ABSTRACT: Amyotrophic lateral sclerosis (ALS) is a progressive degenerative disorder of motor neurons. Most patients who are diagnosed with ALS decline and ultimately perish within 2 to 4 years of symptom onset. There are many symptoms and facets of care that need to be addressed. These include nutrition, respiration, pain management, sialorrhea, muscle cramps and spasticity, vaccinations, fatigue and insomnia, depression and anxiety, and pseudobulbar affect, among others. Multidisciplinary care for these patients is common, and pharmacists have demonstrated they can positively impact patient care for those who have ALS. Examples of drug therapy recommendations and other interventions pharmacists can make are reviewed.
Amyotrophic lateral sclerosis (ALS, or Lou Gehrig’s disease) is a progressive, fatal, degenerative disorder that leads to death of upper and lower motor neurons. The loss of impulses to the brain and spinal cord lead to atrophy of muscle and increased muscle weakness.1 As the bulbar, limb, abdominal, and thoracic muscles progressively weaken, patients experience muscle atrophy, limb spasticity, weight loss, oropharyngeal dysfunction, and the inability to ambulate. Respiratory failure is the usual cause of death in patients, typically within 2 to 4 years after symptom onset.2,3
Etiology/Epidemiology
In 2014, in the United States, 15,927 patients had ALS. This equates to five cases per 100,000 population.4 ALS does not have a known cause; nor is it curable.5,6 There are two types: sporadic ALS (sALS), which comprises 85% to 90% of cases, and familial (fALS), which is less common and occurs in at least two people in the same family.7 Familial ALS is similar between genders, and sALS is more common among men. Advancing age, male gender, and family history are risk factors for the development of ALS. The onset of fALS tends to be earlier compared with sALS.7 Genetic and nongenetic modalities are thought to exist, and gene mapping has identified 30 mutations that play a role in ALS development.7
Neuroprotective and Disease-Modifying Treatments
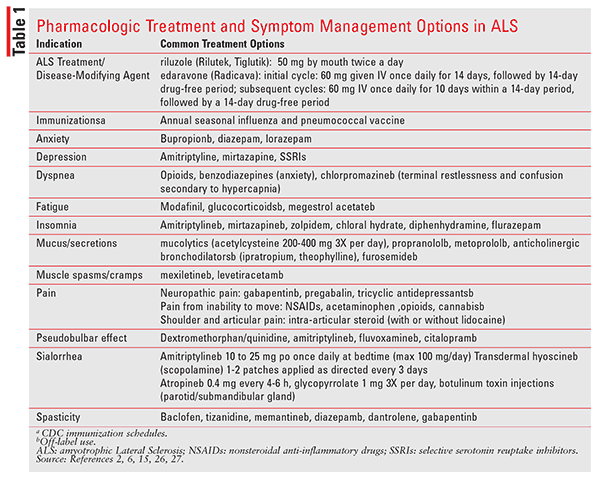
The FDA has approved two medications for ALS treatment—riluzole (Rilutek, Tiglutik) and edaravone (Radicava). The mechanism of action (MOA) of riluzole is unknown; however, its pharmacologic properties include an inhibitory effect on glutamate and voltage-dependent sodium channels. The dose for treatment of ALS is 50 mg twice daily. It should be taken at the same time each day, 1 hour before or 2 hours after a meal. Riluzole is well tolerated; however, nausea and vomiting have been reported. Serum aminotransferases should be monitored at baseline and during therapy for risk of hepatic injury.8,9 Rilutek is a tablet formulation, and Tiglutik is an easy-to-swallow, thickened suspension that is an option for patients with dysphagia.10
The FDA approved edaravone in 2017 for the treatment of ALS. Similar to riluzole, its MOA in treating ALS is unknown. Edaravone prevents oxidative damage to cell membranes and may inhibit the progression of ALS through free-radical scavenging.11 Edaravone is administered intravenously in cycles. Initially, 60 mg is given once daily for 14 days, followed by a 14-day drug-free period. Subsequent cycles are dosed at 60 mg once daily for 10 days within a 14-day period, followed by a 14-day drug-free period. The total infusion of two 30-mg IV bags is consecutively administered over 60 minutes.11 Edaravone contains sodium bisulfite and should be used cautiously in patients with asthma or a sulfite allergy, due to risk of anaphylactic or allergic reaction.11 Other commonly reported adverse reactions include bruising, headache, and abnormal gait.8,11,12
Tested and Emerging Therapies
Riluzole was approved for the treatment of ALS in 1995 and was the only medication on the market until 2017. However, many new therapies are being considered, are under development, or have undergone testing.7 These therapies are placed into categories based on suspected disease pathology. Those groups include anti-inflammatory, antiapoptotic, antiexcitotoxicitory, antiaggregation, antioxidant, neuroprotection and neurotrophic growth factors, stem cell–based therapies, glial-restricted precursors, neural progenitor stem cells, and gene-therapy strategies.7 Despite the advancements in drug development for the treatment of ALS, the mainstay of treatment continues to be symptomatic management.6
Respiratory Management
Respiratory failure is the leading cause of death, mainly due to mechanical failure and inability of the lungs to fully inflate during inspiration. Infection and aspiration may play a role.2,13 Supplemental oxygen is not recommended, except in cases where patients have pulmonary disease or are terminally ill and decline assisted ventilation. Supplemental oxygen in others can hinder patients as it may suppress respiratory drive and lead to carbon dioxide retention and respiratory arrest.13 Cough-assisting devices and chest-wall oscillation are recommended, along with the management of secretions to increase ventilation effectiveness.2 Treatment of dyspnea is addressed later in this article. Pulmonary function should be assessed every 3 months after diagnosis using erect forced vital capacity (FVC) and vital capacity (VC).2,14 The sniff nasal inspiratory force test avoids the use of a mouthpiece and may be a more accurate respiratory assessment for those with bulbar weakness.14 Nocturnal oximetry can be used to identify nocturnal hypoventilation and the need for noninvasive positive-pressure ventilation (NIPPV), as it is usually the earliest sign of respiratory insufficiency.2,6,13,14
NIPPV or invasive mechanical ventilation (IMV) is used to increase survival and decrease symptoms of respiratory insufficiency. Of these, only NIPPV has been documented to increase quality of life (QoL) and is the standard of care for patients with ALS.2,6,13 NIPPV should be introduced when the FVC falls below 50% of predicted; or, if the patient is symptomatic, it can be introduced earlier.13 Initially, NIPPV is only used at night. As respiratory function declines, it can be used in the daytime and then continuously, if needed.25 IMV can be utilized to further extend life. Advance directives and a plan for respiratory insufficiency should be created prior to complications developing.2,6,13 If patients wish to withdraw ventilation, parenteral morphine, a benzodiazepine, and an antiemetic are recommended to reduce dyspnea and anxiety. Paralytic drugs should not be used to aid in ventilator withdrawal.2,6
Nutrition and Dysphagia
Patients with dysphagia may not consume proper calories or fluids, leading to increased muscle atrophy, fatigue, and weakness.6 To prolong survival, percutaneous endoscopic gastrostomy (PEG) placement should be evaluated as dysphagia progresses. Patients with ALS receive assistance from speech pathologists, have their food and fluid consistency modified, are prescribed high calorie and protein supplements, and receive education on diet, swallowing techniques, and postural changes. If these measures fail, then a PEG tube should be placed.2,6 These patients exhibit symptoms such as jaw weakness and fatigue, slow eating, drooling, and choking on fluid and food. PEG placement provides another avenue for oral nutrition and medication administration, and it is the standard of care. Placing a PEG tube, however, may present problems with administration of medications. Pharmacists help counsel patients and caregivers about which medications can be crushed and administered through a PEG tube.
Symptom Management With Pharmacologic Considerations
Sialorrhea (excessive salivation/drooling) is common. Preferred treatments in ALS include off-label use of amitriptyline, oral or transdermal hyoscine, or atropine.2,14,15 Glycopyrrolate may be preferred as first-line therapy due to a lower incidence of central nervous system side effects, such as in patients with cognitive impairment.15 It is not recommended to surgically treat sialorrhea; however, irradiation of the salivary glands can be tried when pharmacologic therapy fails.2,14 Other alternatives for refractory sialorrhea, such as botulinum toxin injections in the parotid and/or submandibular gland, have been effective. 2,14
Thick Bronchial Secretions: Thick mucus is problematic and is a poor prognostic factor in patients with ALS. Providers need to be able to differentiate between this and sialorrhea to select appropriate treatment.2,14 Nonpharmacologic treatments for thick mucus, such as cough augmentation, increased fluid intake, nebulizers with saline, and humidification of air are helpful. Treatment with mucolytics such as acetylcysteine is recommended if sufficient cough flow is present.2,14 Finally, there appears to be some benefit with beta blockers (propranolol, metoprolol), furosemide, and anticholinergic bronchodilators (theophylline, ipratropium).2
Muscle Cramps and Spasticity: Muscle cramps and spasms are quite bothersome in patients with ALS and can cause pain and discomfort, particularly at night.2,14 Quinine sulfate was previously the recommended treatment; however, the FDA has restricted it to treatment of malaria and warned against off-label use due to safety concerns.16 For cramps, levetiracetam and mexiletine have evidence to support their use.2,14 Nonpharmacologic management with exercise, hydrotherapy, and physiotherapy may be helpful.2
Hydrotherapy, heat, cold, electrical stimulation, chemodenervation, and surgery have been utilized to treat muscle spasticity. Physical therapy is the primary treatment for spasticity with positive evidence in ALS.2 Pharmacotherapy with baclofen and tizanidine are typically recommended. Intrathecal baclofen is usually avoided, but it can be utilized in severe cases if oral therapy is insufficient or in patients with slow progression and primarily upper-motor impairment.2,13,14 Additional therapies that can be considered include memantine, dantrolene, diazepam, tizanidine, and gabapentin.2
Depression and anxiety are common among patients with ALS, with estimates of depression ranging from 4% to 56%.14,17 Reported rates of depression in ALS tend to be higher than depression in the general population.14 Regardless of the prevalence, depression plays a significant role in the QoL of those with ALS and should be recognized. No formal, controlled studies have explored the treatment of depression or anxiety in ALS. Antidepressants are recommended in appropriate patients, and the choice of an agent may be determined by presence of other ALS symptoms (sialorrhea, apathy, appetite loss, insomnia).2 Amitriptyline, mirtazapine, and selective serotonin reuptake inhibitors are commonly used for treatment of depression.2,14 Antidepressants with anxiolytic effects could be used for treatment of anxiety; however, other therapy is commonly required. Bupropion and benzodiazepines, such as diazepam suppositories and tablets, or lorazepam are recommended.2
Pseudobulbar Affect, also referred to as emotional lability, is a condition that is marked by episodes of uncontrolled crying or laughing which is inappropriate or of disproportionate intensity and that is associated with various neurological disorders.18 Pseudobulbar affect can be socially troubling and negatively impact a patient’s QoL. It occurs in approximately 50% of patients with ALS and is therefore an important treatment consideration.2,6 In 2010, the FDA approved the first medication labeled for treatment of pseudobulbar affect, dextromethorphan and quinidine (D/Q, Nuedexta).19 The dosage recommendations for D/Q 20 mg/10 mg is one capsule by mouth once daily for 7 days, followed by one capsule twice daily. It is recommended to periodically reassess for continued use and to avoid exceeding D/Q 40 mg/20 mg in a 24-hour period.14,19 The most common adverse reactions are diarrhea, dizziness, peripheral edema, vomiting, cough, and weakness.19 Although the dose of quinidine in this preparation is only 1% to 3% of those needed to treat arrhythmias, cardiac monitoring needs to be conducted (QT interval assessed at baseline, within 3-4 hours after the first dose for patients at risk of QT prolongation). During and prior to therapy, magnesium, potassium, CBC, and liver and renal function should be assessed.19
Aside from this FDA-approved treatment for pseudobulbar affect, off-label treatment with amitriptyline, fluvoxamine, or citalopram can be considered.2,6,14 Amitriptyline is a good choice for patients who have sialorrhea and pseudobulbar affect, as it treats both symptoms and helps to simplify the medication regimen.
Fatigue and Insomnia: Fatigue in ALS can be a result disease-modifying therapy, or it can result from effort required to conduct daily living activities.14 Some studies demonstrated improvement in fatigue with modafinil, glucocorticoids, and megestrol acetate.2,14
Insomnia can develop in ALS patients as a result of depression, nonobstructive hypoventilation, central apneas, restless leg syndrome, anxiety, excessive secretions, increased myoclonic activity, difficulty changing position due to muscle weakness, cramps, pain, dysphagia, dyspnea, respiratory distress, or other causes, especially in the final months of life.2,14 Similar to other patients who are having sleep difficulties, the underlying cause should be identified and treated, and medications should be used sparingly and only when appropriate. Recommendations for treatment of insomnia in ALS include the use of amitriptyline, mirtazapine, zolpidem, chloral hydrate, diphenhydramine, and flurazepam, with the most common being amitriptyline and zolpidem.2,14
Nonpharmacologic Symptom Management
Pharmacists, as members of a multidisciplinary team providing care to ALS patients, should be familiar with nonpharmacologic symptom management. For example, for muscle weakness and functional decline, pharmacists could recommend or carry the following products: specialized eating utensils and holders, pressure-relieving mattresses to prevent pressure ulcers, removable headrests, bathtub lifts, higher toilet seats, canes, crutches, walking frames, wheelchairs, and ankle/foot orthoses.14 Patients with ALS may have difficulty communicating from dysarthria. Augmentive communication systems can be used for communication and prosthetic treatments can help with articulation and reducing hypernasality. Finally, referral to a speech and language therapist could be recommended.14
Venous Thromboembolism Prevention
A higher incidence of venous thromboembolism (VTE) has been found in ALS patients when compared with controls, due to impairment in mobility and respiratory function.20-22 The estimated annual incidence of VTE in the ALS population has been found to range between 3% and 11.2%, and in those with leg-onset ALS or significant leg weakness, the incidence was between 35.8% and 35.5%, respectively.22,23 Without additional risk factors, there are no recommendations to prophylactically treat patients with ALS with anticoagulation.2,24 Pharmacists should familiarize themselves with treatment recommendations for VTE in the event one would develop. They can also ensure that patients with ALS are optimally managing risk factors and taking measures such as the use of compression stockings, limb elevation, and physiotherapy.2
Vaccine Considerations
Patients with ALS should receive the annual influenza vaccine and pneumococcal vaccine.13,14 Immunization guidelines recommend the influenza vaccine for those with neurologic conditions that can compromise handling of respiratory secretions. Additionally, in those with chronic pulmonary conditions, it is recommended that they receive the pneumococcal vaccine
End-of-Life Care
When patients enter the terminal phase of the disease, the focus shifts from maximizing function to providing palliative care.6 Patients should be referred to hospice in the terminal stage of their disease, as it has proven beneficial. End-of-life issues should be discussed well in advance of the terminal phase, and options, including respiratory support, should be reevaluated every 6 months.2,6 Palliative-care physicians are becoming increasingly more involved in providing care to patients with ALS.13 They may provide advice on diet, supplements, exercise, and common medication interventions.13 Pharmacists, working as part of an interdisciplinary team, could take the lead and help to make some of these recommendations. Pain, dyspnea, and anxiety are among the top treatment considerations in terminal ALS patients.6
Dyspnea, particularly in the terminal stage of ALS, is problematic as a result of respiratory muscle weakness.6 To treat dyspnea, opioids are recommended in combination with benzodiazepines if the patient is also experiencing anxiety.2 Reversible causes of dyspnea should be considered and treated if present.6 If the patient has terminal restlessness and confusion, chlorpromazine should only be used.2 Oxygen alone should be used to treat symptomatic hypoxia.2,6
Pain: Significant pain is something that most individuals with ALS experience, particularly later in the disease.6,13,25 Pain is likely due to immobility, stiff joints, muscle cramps, itching, pressure on the skin or joints, mechanical back pain, neuropathic pain, spasticity, comorbid conditions, facial pressure ulcers from the noninvasive ventilation mask, suctioning of saliva, neck pain, and adhesive capsulitis.6,13,14,25 Due to the different locations and causes, treatment modalities have been proposed, including nonsteroidal anti-inflammatory drugs, neuropathic medications, cannabinoids, and opioids.6,14,25 Few studies of pain in patients with ALS have been conducted.14,25 Additionally, until about 10 years ago, pain was been an overlooked by providers as a symptom among ALS patients.25 Pain is associated with decreased QoL and increased risk of depression. Of note, it appears that the presence of pain, rather than its severity, plays a large role in the distress that ALS patients experience.25 Providers are aware of the importance of pain management; however, it has been reported that there is a need for additional training and use of standard protocols for assessing pain.25 Pharmacists, as medication experts, could fill in this gap on multidisciplinary teams by inquiring about the source and intensity of pain with appropriate tools, providing pain education, and making recommendations for treatment based on its source and intensity. Considerations for the treatment of pain and other ALS symptoms can be found in TABLE 1.
Additional Role of the Pharmacist
The role of a pharmacist in a multidisciplinary ALS clinic has been evaluated.3 Clinical pharmacists provided education on 2.5 topics, made an average of two interventions, and spent an average of 21 minutes of face-to-face contact per patient.3 The pharmacist in this setting provided education and discussed dietary supplements; administration of medications through a feeding tube; and the dosing, efficacy, and side effects of medications. While there are few interventional therapies used for the treatment of ALS, pharmacists can provide medication optimization, medication-related education, and discussion of general medication issues.3
Pharmacists in hospitals will have a role in pharmacy and therapeutics considerations for existing therapies and those entering the market in the near future. In addition, they may need to provide instructions for patients who will be navigating the prior authorization (PA) process in the outpatient setting. PAs for edaravone and riluzole may state that patients must meet the inclusion criteria of trials that permitted their FDA approval.26 Finally, some PAs may require failure with riluzole or its concomitant use.26 Pharmacists can work with prescribers to select appropriate therapy for the patient and ensure it will be covered by insurance.
Conclusion
ALS is a complex disease with multiple symptoms that must be managed. Pharmacists are in a prime position to familiarize themselves with the progression of the disease and symptoms patients may experience. They can then utilize their expertise to provide pharmacologic and nonpharmacologic treatment recommendations for the patient based on their presentation as part of a multidisciplinary care team.
REFERENCES
1. Amyotrophic lateral sclerosis (ALS). Cleveland Clinic, The Cleveland Clinic Foundation. https://my.clevelandclinic.org/health/diseases/16729-amyotrophic-lateral-sclerosis-als. Accessed May 31, 2019.
2. Andersen PM, Abrahams S, Borasio GD, et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)–revised report of an EFNS task force. Eur J Neurol. 2011;19(3):360-375.
3. Jefferies KA, Bromberg MB. The role of a clinical pharmacist in a multidisciplinary amyotrophic lateral sclerosis clinic. Amyotroh Lateral Scler. 2012;13(2):233-236.
4. Mehta P, Kaye W, Raymond J, et al. Prevalence of amyotrophic lateral sclerosis - United States, 2014. MMWR Morb. 2018;67(7):216-218.
5. Amyotrophic lateral sclerosis (ALS). Mayo Clinic, Mayo Foundation for Medical Education and Research. www.mayoclinic.org/diseases-conditions/amyotrophic-lateral-sclerosis/symptoms-causes/syc-20354022. Accessed July 23, 2019.
6. Miller RG, Jackson CE, Kasarskis EJ, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidenced-based review). Neurology. 2009;73:1218-1226.
7. Nowicka N, Juranek J, Juranek JK, Wojtkiewicz J. Risk factors and emerging therapies in amyotrophic lateral sclerosis. Int J Mol Sci. 2019;20:2616.
8. Roberson ED. Treatment of central nervous system degenerative disorders. In: Brunton LL, Hilal-Dandan R, Knollmann BC, eds. Goodman & Gilman’s: The Pharmacological Basis of Therapeutics. 13th ed. New York, NY: McGraw-Hill. http://accesspharmacy.mhmedical.com.ezproxy.findlay.edu:2048/content.aspx?bookid=2189§ionid=169518826. Accessed June 1, 2019.
9. Lexicomp [Internet]. Hudson, OH: Wolters Kluwer. 2019. https://online.lexi.com/lco/action/home. Accessed June 1, 2019.
10. ITF Pharma. ITF Pharma announces FDA approval of Tiglutik (riluzole) oral suspension for the treatment of amoytrophic lateral sclerosis (ALS). https://itfpharma.com/wp-content/uploads/2018/09/FINAL-FOR-FDA-FILING_US-FDA-Approves-TIGLUTIK-Press-Release_TIGLPA092018.pdf. Accessed June 2, 2019.
11. Lexicomp[ Internet]. Hudson,OH: Wolters Kluwer. 2019. https://online.lexi.com/lco/action/home. Accessed June 2, 2019.
12. Radicava [package insert]. Jersey City, NJ: Mitsubishi Tanabe Pharma Corporation; 2017.
13. Karam CY, Paganoni S, Joyce N, et al. Palliative care issues in amyoamyotrophic lateral sclerosis: an evidenced-based review. Am J Hosp Palliat Care. 2016;331(1):84-92. 14. Galvaz-Jiminez, N. Symptom-based management of amyotrophic lateral sclerosis. Up to Date, 16 May 2019. www.uptodate.com/con-tents/symptom-based-management-of-amyotrophic-lateral-sclerosis#! Accessed May 31, 2019. 15. Garuti G, Rao F, Ribuffo V, Sansone VA. Sialorrhea in patients with ALS: current treatment options. Degen Neurol Neuromuscul Dis. 2019;9:19-26.16. Food and16. Pseudobulbar affect. In: Merriam Webster. [online]. www.merriam-webster.com/medical/pseudobulbar%20affect. Accessed June 2, 2019.17. Atassi N, Cook A, Pineda CM, et al. Depression in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2011;12(2):109-112.
18. Pseudobulbar affect. In: Merriam Webster. [online]. www.merriam-webster.com/medical/pseudobulbar%20affect. Accessed June 2, 2019.
19. Lexicomp [Internet]. Hudson, OH: Wolters Kluwer. 2019. Nuedexta. https://online.lexi.com/lco/action/home. Accessed June 1, 2019.
20. Kimura F, Qureshi M, Cudkowicz M, et al. Increased incidence of deep venous thrombosis. ALS Neurology. 2007;68(23):2046-2047.
21. Kupelian. April 09, 2019; 92(15 Supplement). May 8, 2019.
22. Elman LB, Siderowf A, Houseman G, et al. Venous thrombosis in an ALS population over four years. Amyotroph Lateral Scler Other Motor Neuron Disord. 2005;6(4):246-249.
23. Gladman M, Dehaan M, Pinto H, et al. Venous thromboembolism in amyotrophic lateral sclerosis: a prospective study. Neurology. 2014;82(19):1674-1677.
24. Schunemann HJ, Cushman M, Burnett AE, et al. American Society of Hematology 2018 guidelines for management of venous thromboembolism: prophylaxis for hospitalized and nonhospitalized medical patients. Blood Adv. 2018;2:3198-3225.
25. Chio A, Mora G, Lauria G. Pain in amyotrophic lateral sclerosis. Lancet Neurol. 2017;16:144-157.
26. Santaniello B. ALS managed care considerations. Am J Manag Care. 2018;24(15 Suppl):S336-S341.


