US Pharm. 2017;42(5)(Specialty&Oncology suppl):28-31.
ABSTRACT: Hypertension, specifically pulmonary hypertension, is a syndrome that can affect pediatric patients as well as adults. Treatment can be challenging because of the lack of FDA-approved medications and tolerable dosage forms for pediatric patients. Current classes of medications primarily used to treat pediatric hypertension include phosphodiesterase inhibitors, endothelin receptor antagonists, and prostacyclins. Additional agents that may be utilized in selected pediatric patients include calcium channel blockers, anticoagulants, and inhaled nitric oxide. Pharmacists should be familiar with the available therapies when caring for pediatric patients with pulmonary hypertension.
Pulmonary hypertension (PH) in pediatric patients, while rare, can be a life-threatening condition. There is no cure for PH, only treatment options for children that are largely based on the results of adult studies. These therapies, however, can improve quality of life and survival. Pediatric PH is defined as a resting mean pulmonary artery pressure (mPAP) 5 mmHg (at sea level) in term infants after the age of 3 months.1 Pulmonary arterial hypertension (PAH) is a subclassification of PH in which patients have a normal pulmonary capillary wedge pressure of 15 mmHg.1 It is important that pharmacists be familiar with the available therapies when caring for pediatric patients with PH.
PATHOPHYSIOLOGY
In pediatric PH, blood flow exiting the right side of the heart faces greater resistance due to increased muscle present in the walls of the lungs. The right ventricle then enlarges and thickens in response, which may lead to heart failure.2 PH may be related to lung, cardiac, or systemic diseases such as congenital abnormalities of the heart; lung diseases related to prematurity; chronic thromboembolic disease; or diseases that primarily affect other organs, including scleroderma, liver diseases, hereditary hemorrhagic telangiectasia, AIDS/HIV, and sickle cell disease.3
PAH that exists in the absence of a classifiable cause or associated underlying disease state is referred to as idiopathic pulmonary arterial hypertension (IPAH). IPAH is a diagnosis of exclusion; specifically, the absence of diseases of the left side of the heart or valves, lung parenchyma, thromboembolism, or other causes. Heritable pulmonary arterial hypertension (HPAH) is PAH that is associated with positive family or genetic evaluation. While genetic tests are not routinely used to guide the management of patients with PAH, genetic testing may be offered to any individual with a family history of PAH or IPAH.4,5
Pediatric PH may be associated with impaired functional and structural adaptation of the pulmonary circulatory system during the developmental period from fetal to postnatal life. The most common symptoms of PH are related to difficulty breathing and may include progressive shortness of breath, fatigue, hyperventilation, progressive weakness, lightheadedness or dizziness, fainting spells, or progressive cyanosis.
At the time of initial PH diagnosis, an inclusive history and physical examination, diagnostic testing for the assessment of PH pathogenesis and classification, and an assessment of cardiac function should be performed. Diagnostic tests include chest x-ray, electrocardiogram, echocardiogram, chest computed tomography, laboratory studies, exercise capacity test, and cardiac catheterization.4
TREATMENT
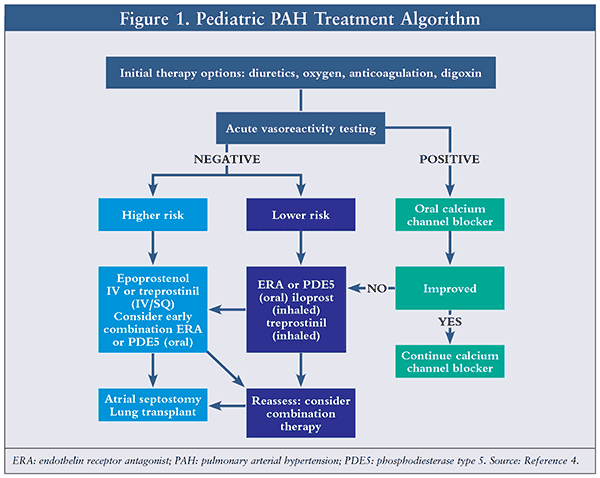
Many medications have been used and studied in the treatment of pediatric PAH. However, there are three medication classes that have been evaluated more thoroughly for their efficacy in pediatric PH treatment: phosphodiesterase type 5 (PDE5) inhibitors, endothelin (ET) receptor antagonists, and prostacyclin agonists. Other medications used are calcium channel blockers, anticoagulants, and inhaled nitric oxide (iNO). Prior to initiation of targeted PH therapy, the patient should be assessed for acute vasodilator responsiveness via cardiac catheterization; left-sided heart disease or pulmonary venous disease resulting in an anatomic obstruction should be excluded. Medication therapy is determined based on patient responsiveness to acute vasodilator testing (AVT; Figure 1). AVT is used to assess the response of the pulmonary vascular bed to pulmonary-specific vasodilators. In children with IPAH or isolated pulmonary hypertensive vascular disease, response to AVT is defined as a decrease in mPAP of at least 10 mmHg to <40 mmHg, with normal or increased cardiac output and a decrease in mPAP 20%; an increase or no change in cardiac index; and a decrease or no change in pulmonary vascular resistance/systemic vascular resistance ratio.4
Phosphodiesterase Inhibitors
In patients with PH, PDE5 expression and activity are increased. PDE5 inhibitors promote pulmonary vascular dilation and remodeling, as well as increase smooth-muscle cyclic guanosine monophosphate levels. They are recommended for use in patients who have tested negative on AVT. PDE5 inhibitors are commonly administered orally, with good tolerability. Adverse effects most frequently seen are agitation, flushing, and headache. Although rare, ocular effects include transient blue/green visual abnormalities and blurred vision; changes in light perception may occur.1
Sildenafil: Sildenafil has been approved for treatment of adult PAH and has been studied in pediatric patients. The early STARTS-1 trial, which examined varying oral sildenafil doses in pediatric PAH, found positive outcomes on measured parameters such as functional capacity (high dose), mPAP (medium dose), and pulmonary vascular resistance index (PVRI; medium and high dose).6 However, the continued trial, STARTS-2, showed an increased risk of mortality in patients randomized to high-dose sildenafil in the STARTS-1 trial and in patients initially randomized to placebo and then switched to high-dose sildenafil. After 3 years, the patients in the high-dose sildenafil group had a mortality rate of 20%, while those in the medium-dose group had a mortality rate of 14%. Furthermore, older patients with IPAH who had higher mPAP and PVRI upon enrollment were found to have the greatest risk of mortality.
Of note, patients with PAH associated with congenital heart disease and patients who weighed <20 kg had a different mortality risk compared with the other children in the study.7 As a result of the STARTS-1 and STARTS-2 trials, the FDA issued a warning in 2012 against the chronic use of sildenafil in patients aged 1 to 17 years.8 In light of concern about the study design and “lack of correlation to clinical practice,” the FDA updated the warning in 2014 to state that “health care professionals must consider whether the benefits of treatment with the drug are likely to outweigh its potential risks for each patient.”9,10
Other recommendations suggested cautious use of oral sildenafil, avoiding high-dose therapy. For patients aged <1 year, the recommended dose of sildenafil is 0.5-0.1 mg/kg three times daily orally.4 Alternatively, the recommended dosage for patients <20 kg is 10 mg three times daily orally, and for patients >20 kg, it is 20 mg three times daily orally.1,4 Sildenafil is available in varying tablet strengths and a suspension, and it is the only available IV PDE5 inhibitor. IV sildenafil is used only in an acute-care setting because of its risk of systemic effects.1,11
Tadalafil: Tadalafil is a longer-acting PDE5 inhibitor that has been shown to decrease clinical worsening and improve exercise capacity and health-related quality of life in adult patients who have PAH or IPAH. Because of the drug’s documented efficacy and the sildenafil warnings, there has been an increase in pediatric use of tadalafil. Though tadalafil is not FDA-approved in pediatric patients, a retrospective study showed an initial dosage of 1 mg/kg/day is tolerated well by pediatric patients, demonstrates clinical improvement, and has a favorable side-effect profile.12 Tadalafil is available in a tablet that can be extemporaneously compounded into an oral suspension. Use of tadalafil is contraindicated in neonates and infants because of their immature metabolic pathways. Adverse reactions seen with tadalafil are similar to those seen with sildenafil, but tadalafil has little PDE6 inhibition and therefore is associated with fewer visual effects.1,4
Endothelin Receptor Antagonists
ET-1 is a potent vasoreactive peptide, primarily in the vascular endothelial and smooth-muscle cells, and is the major ET associated with PAH.2 ET-1s effects are mediated through receptor subtypes ETA and ETB, which mediate vasoconstriction in vascular smooth muscle.1,2 ET receptor antagonists (ERAs) are associated with improved hemodynamics and survival in adult PAH patients and have been studied in pediatric patients. Common adverse effects of ERAs include elevated liver enzymes, peripheral edema, and anemia; additionally, ERAs are known teratogens, and male infertility has been reported. Despite clinical benefits seen in pediatric patients with IPAH or associated PAH, ERAs are not FDA-approved for use in pediatric patients.1,4 Access to ERAs is restricted to registered pharmacies/pharmacists, due to the requirement of the Risk Evaluation and Management Strategy program.
Bosentan: Bosentan is a dual ERA that improves exercise capacity in adults with PAH, as well as lowering PAP and pulmonary vascular resistance; similar results have been seen in pediatric patients. Children with IPAH or PAH associated with congenital heart disease typically tolerate bosentan well. Compared with adults, children had a lower incidence of liver-enzyme elevation and a lower medication-discontinuation rate. Based on the available trials, doses of 31.25 mg, 62.5 mg, or 125 mg twice daily are recommended for therapy in pediatric patients with PAH weighing 10 to 20 kg, >20 to 40 kg, or >40 kg, respectively. If the patient weighs <10 kg, the maintenance dosage is 2 mg/kg twice daily orally. Bosentan is available as a 62.5- and a 125-mg tablet that is often split; it may be dissolved in water for use in pediatric patients. Long-term monitoring of liver function tests is required with bosentan.1,4
Ambrisentan: Ambrisentan is an orally administered selective ETA receptor antagonist. This drug may benefit patients with PAH by blocking the vasoconstrictive properties of the ETA receptors while keeping the vasodilatory and clearance properties of ETB receptors.1 In a retrospective study of 38 pediatric PAH patients, the mPAP and functional class improved in patients treated with ambrisentan as replacement therapy for bosentan or as add-on therapy with bosentan.13 Despite the lack of studies conducted on ambrisentan in children with PAH, use is increasing due to decreased the risk of elevated liver enzymes, lack of drug-drug interaction with PDE5 inhibitors, and once-daily dosing. A current retrospective study showed initial doses of 2.5 mg or 5 mg may be used in pediatric patients weighing <20 kg or >20 kg, respectively.2 Liver function tests do not need to be monitored regularly in patients using ambrisentan, unlike those taking bosentan.1,4
Prostacyclins
In children with congenital heart disease and adults with IPAH and HPAH, there is an imbalance in the biosynthesis of prostacyclin and thromboxane A2. This imbalance favors thromboxane A2 synthesis and vasoconstriction. Prostacyclins increase pulmonary vasodilation through stimulation of the adenosine cyclic monophosphate pathway. Prostacyclins are considered the gold standard of treatment, as they have been associated with improved outcomes in adult and pediatric patients.1
Epoprostenol: Epoprostenol was the first prostacyclin approved by the FDA for use in adults. In children and adults, survival and quality of life are improved with the long-term use of IV epoprostenol.14,15 Due to its short half-life, epoprostenol must be infused at a starting dosage of 1 to 3 ng/kg/minute. A continuous infusion must be initiated in the hospital, and the dose can be steadily increased every 1 to 2 weeks, as tolerated. Catheter complications are common and the dose should not be acutely discontinued due to the potential for rebound hypertension. Dose-dependent adverse reactions such as rash, nausea, jaw pain, headaches, bone pain, or thrombocytopenia may occur.1,4
Treprostinil: Treprostinil is a prostacyclin that is FDA-approved in adults for use subcutaneously, orally, and intravenously. IV treprostinil outcomes and hemodynamic changes in treated children have been shown to be similar to those of epoprostenol, with fewer adverse reactions. The dose depends on the route of administration, but many children have been successfully switched from epoprostenol to IV treprostinil.1 Inhaled treprostinil has been shown to be as effective as iNO in lowering PAP and PVRI. It may be initiated in the outpatient setting as add-on therapy in patients who are stable. Common adverse reactions of inhaled treprostinil are throat irritation, nausea, cough, flushing, and headache. Site discomfort may be seen with the subcutaneous injection, but may be relieved by administration of a systemic histamine blocker, oral analgesic, or application of a topical analgesic. Of note, catheter-related bloodstream infections have increased in patients with PAH treated with IV treprostinil. Oral dosing of treprostinil has not been fully evaluated in children.1,4
Iloprost: Iloprost is a prostaglandin approved for inhalation treatment of adult PAH. While no pediatric dose has been established, the drug has been administered to children through a nebulizer six to nine times per day.4 Because of the treatment duration and frequency, it is often difficult to administer iloprost to young children. In some children, iloprost administration has resulted in an increase in airway reactivity. Iloprost has also been studied in conjunction with medications such as sildenafil and bosentan, but efficacy has not been clearly defined in children.1,4
Calcium Channel Blockers
Patients who qualify for an acute trial of oral calcium channel blocker (CCB) therapy are those who have previously been acutely reactive to iNO (positive AVT) or IV epoprostenol. Recommended medications for long-term CCB therapy include nifedipine 2 to 5 mg/kg/day; diltiazem 3 to 5 mg/kg/day; and amlodipine 2.5 to 10 mg/day.1 These medications, especially diltiazem, may lower the heart rate and can cause a significant decrease in blood pressure or cardiac output. Because of this, using CCBs to evaluate vasoreactivity comes with significant risks. Careful monitoring of patients is essential due to the potential for patient deterioration over time with CCB monotherapy.1 Additionally, administration in pediatric patients can pose challenges, particularly with the capsule/extended-release-only forms of nifedipine; diltiazem and amlodipine are available in tablets that can be crushed. Amlodipine and nifedipine may also be extemporaneously compounded.
There are several contraindications for short- or long-term CCB therapy in pediatric patients. CCB therapy is contraindicated in children with low cardiac output, elevated right atrial pressure, or right ventricular failure; who are nonresponders to iNO or IV epoprostenol; or who have not undergone ATV. CCBs are not recommended in children aged <1 year, because of the tendency toward more prominent negative inotropic effects in this age group. Of note, verapamil is contraindicated in patients with PAH due to its negative inotropic effects, tendency to cause bradycardia, and minimal pulmonary vasoreactive properties.1,4
Anticoagulation
In children with PAH, the benefits of long-term anticoagulation have not been studied. Despite the lack of data, however, it is still recommended to use anticoagulants (with warfarin being the drug of choice in adults) in patients with IPAH/HPAH, long-term indwelling IV catheters, low cardiac output, and/or hypercoagulable states. Studies have shown survival improvement in adult PAH patients treated with anticoagulants.16,17 The risk/benefit ratio should be strongly considered prior to initiating anticoagulant therapy, particularly in small children susceptible to hemorrhagic complications. Other anticoagulants, including aspirin, have not been studied for their use in this patient population.4
Inhaled Nitric Oxide
iNO is a gas that induces vasodilation and improves oxygenation; it is commonly used in the acute treatment of PAH. iNO is approved by the FDA for use as a specific pulmonary vasodilator therapy for persistent PH of the newborn; it may also be used in infants with established bronchopulmonary dysplasia. Despite its common use, data on efficacy are not available; it has been shown that iNO does not reduce hospitalization length or mortality in some patients. Therapy is often started at a dosage of 10 to 20 parts per million (ppm), with subsequent weaning to 2 to 10 ppm. Due to its short half-life, risk of toxic metabolites, and complicated delivery system, iNO has not been used as long-term therapy for PAH management.1,4
CONCLUSION
Many medications have been studied and successfully used in the treatment of pediatric PH. Although only iNO has been approved by the FDA for use in pediatric patients, each drug has shown benefits for these patients in clinical trials. Pharmacists can provide integral care for these patients by closely monitoring for clinical worsening and adverse reactions and assessing the feasibility of available therapies.
REFERENCES
1. Voehies EE, Ivy DD. Drug treatment of pulmonary hypertension in children. Paediatr Drugs. 2014;16(1):43-65.
2. D’Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension: results from a national prospective registry. Ann Intern Med. 1991;115:343-349.
3. Barst RJ, McGoon MD, Elliott CG, et al. Survival in childhood pulmonary arterial hypertension: insight from the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Circulation. 2012;125:113-122.
4. Abman SH, Hansmann G, Archer SL, et al. Pediatric pulmonary hypertension: guidelines from the American Heart Association and American Thoracic Society. Circulation. 2015;132(21):2037-2099.
5. Simonneau G, Galie N, Rubin LJ, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 suppl):D34-D41.
6. Barst RJ, Ivy DD, Gaitin G, et al. A randomized, double-blind, placebo-controlled, dose-ranging study of oral sildenafil citrate in treatment-naïve children with pulmonary arterial hypertension. Circulation. 2012;125(2):324-334.
7. Barst R, Baghetti M, Pullido T,et al. STARTS-2: long-term survival with oral sildenafil monotherapy in treatment-naïve pediatric pulmonary arterial hypertension. Circulation. 2014;129(19):1914-1923.
8. FDA. FDA Drug Safety Communication: FDA recommends against use of Revatio (sildenafil) in children with pulmonary hypertension. August 30, 2012. www.fda.gov/Drugs/DrugSafety/ucm317123.htm. Accessed April 3, 2017.
9. Hopper RK, Abman SH, Ivy DD. Persistent challenges in pediatric pulmonary hypertension. Chest. 2016;150(1):226-236.
10. FDA. Revatio (sildenafil). Drug Safety Communication - FDA clarifies warning about pediatric use for pulmonary arterial hypertension. March 31, 2014. www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm391152.htm. Accessed April 3, 2017.
11. Abman SH, Kinsella JP, Rosenzweig EB, et al. Implications of the U.S. Food and Drug Administration warning against the use of sildenafil for the treatment of pediatric pulmonary hypertension. Am J Respir Crit Care Med. 2013;187(6):572-575.
12. Takatsuki S, Calderbank M, Ivy DD. Initial experience with tadalafil in pediatric pulmonary arterial hypertension. Pediatr Cardiol. 2012;33(5):683-688.
13. Takatsuki S, Rosenzweig EB, Zuckerman W, et al. Clinical safety, pharmacokinetics, and efficacy of ambrisentan therapy in children with pulmonary arterial hypertension. Pediatr Pulmonol. 2012;48(1):27-34.
14. McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension. JACC. 2009;53(17):1573-1619.
15. Yung D, Widlitz AC, Rosenzweig EB, et al. Outcomes in children with idiopathic pulmonary arterial hypertension. Circulation. 2004;110(6):660-665.
16. Fuster V, Steele PM, Edwards WD, et al. Primary pulmonary hypertension: natural history and the importance of thrombosis. Circulation. 1984;70:580-587.
17. Frank H, Mlczoch J, Huber K, et al. The effect of anticoagulant therapy in primary and anorectic drug-induced pulmonary hypertension. Chest. 1997;112:714-721.


