US Pharm. 2024;49(2):18-24.
ABSTRACT: In less than 1 year, the FDA has approved four new agents for rare diseases, a class of conditions that affect fewer than 20,000 people in the country. The development of drugs for rare diseases has been fostered by the 1983 Orphan Drug Act and the Rare Disease Cures Accelerator-Data and Analytics Platform. In August 2023, palovarotene (Sohonos) and pozelimab-bbfg (Veopoz) were approved for fibrodysplasia ossificans progressiva and CHAPLE disease, respectively. A month later, nedosiran (Rivfloza) was approved for primary hyperoxaluria type 1. Lastly, givinostat (Duvyzat) was approved in March 2024 for the treatment of Duchenne muscular dystrophy.
The Genetic and Rare Diseases (GARD) center, a subentity of the National Institutes of Health, recognizes more than 10,000 rare diseases.1 The term rare disease is utilized to denote a condition that affects a small portion of the population; for this reason, the definition will vary based on the country or region.2 In the United States, a rare disease is a disease or condition that affects fewer than 20,000 people.1 Rare diseases, also known as orphan diseases, were brought to mainstream attention in the early 1980s through the works of a coalition that evolved into the National Organization for Rare Disorders.3 Together, they lobbied for the passage of the 1983 Orphan Drug Act, which incentivized the medical and pharmacological development of treatments by offering a 7-year period of market exclusivity for a drug approved to treat an orphan disease and tax credits of up to 50% for research and development expenses. A further incentive was the introduction of orphan status by the FDA, which comes with grants for clinical testing and assistance in protocol and investigation design.
Since the enactment of the 1983 Orphan Drug Act until 2022, 882 drugs have received initial approval, which represents 14% of drugs that have received orphan drug designations.4 Collectively, this represents treatment for 392 diseases. Cancers account for close to 60% of the top 25 most designated diseases. Drug development for rare diseases is hindered by many challenges, including lack of disease state information; limited patient population, which complicates trial design and generalizability; and unclear clinically significant outcomes.5 In an effort to overcome such challenges, the Rare Disease Cures Accelerator-Data and Analytics Platform (RDCA-DAP) was launched in 2019, and as of December 2022, it hosts 64 datasets representing more than 24 disease states. The continued efforts of researchers and rare disease advocates have been met with success, as seen by the four novel agents that were approved from August 2023 to April 2024 for four unique rare diseases. This article will briefly review each disease state, general management principles, and the corresponding novel agent.
Fibrodysplasia Ossificans Progressiva
Disease Overview: Fibrodysplasia ossificans progressiva (FOP) is one of three diseases that cause heterotopic ossification (HO), or extraskeletal bone formation.6,7 Characterized by HO in muscles, tendons, and ligaments, FOP is a progressive disease affecting about one per 1.5 million to 2.0 million people.8 In 2006, this ultrarare disease was linked to a genetic missense mutation in the ACVR1 (activin receptor type 1) gene. In most patients with FOP, this genetic mutation arises spontaneously; however, there have been reports of autosomal-dominant inheritance from a parent. The genetic mutation has been connected to ligand-dependent and -independent activation of the bone morphogenetic protein (BMP) signaling cascade, which leads to abnormal chondrogenesis or osteogenesis.
The clinical presentation and progression of the disease are unique for each patient.9 Commonly, patients with FOP will be born with malformations of the great toes, such as hallux valgus and macrodactyly.8 The next stage of the disease is traditionally marked by flare-ups, characterized by sporadic episodes of soft tissue swelling associated with pain and warmth, that occur within the first decade of life (around age 5 years). These episodes may be triggered by trauma or occur spontaneously and are classically the precursor to HO formation. Joints that are typically first affected include the cervical and thoracic spine, thereby limiting neck mobility. The progressive ossification of joints in a proximal to distal and dorsal to ventral pattern inevitably restricts movement, making patients wheelchair bound by their late 20s, and is associated with a range of complications, including thoracic insufficiency syndrome, malnutrition, and deep vein thrombosis formation.
General Management: Until recently, treatment of FOP has been focused on preventative measures and symptomatic management of flare-ups.9 The goal of preventative measures is to decrease the risk of injury and illness to reduce local and systemic inflammation.8-10 Early recognition of the signs of FOP will prevent harmful biopsies of the newly forming HO, which accelerate the inflammatory-mediated ossification and are unnecessary, as diagnosis is made through genetic testing. Patients are educated about mitigating their risk of soft-tissue injury by not participating in high-risk sports, eliminating extrinsic fall risks (proper footwear, assistive devices, environmental hazards), adapting bedding or seating to reduce pressure on bony protrusions, and intramuscular injections (e.g., vaccines). Due to the risk of jaw ankylosis, dental care should be done routinely but cautiously; once jaw movement becomes restricted, the patient should be referred to a dietician to reduce the risk of malnutrition. Decline of pulmonary function can be delayed through performing routine respiratory exercises, such as singing and spirometry.
With no medications specifically indicated for the treatment of FOP until recently, pharmacotherapy was used for the prevention and treatment of flare-ups, often based on expert opinion and not on clinical studies.10 Corticosteroids and nonsteroidal anti-inflammatory drugs are traditionally the mostly widely used agents, but other medications, including mast cell stabilizers, leukotriene inhibitors, bisphosphonates, tyrosine kinase inhibitors, and muscle relaxants, have anecdotal evidence to support their use. The International Clinical Council on FOP and consultants released FOP treatment guidelines in 2022 that provide guidance on dosing and duration of medication use (summarized in TABLE 1).11 An additional statement was released in March 2024 regarding the use of canakinumab, tofacitinib, and imatinib.12 These agents are only recommended when conventional therapy for severe and intractable flare-ups has failed or for patients with severe/rapid progression of disease who have no contraindications to use.
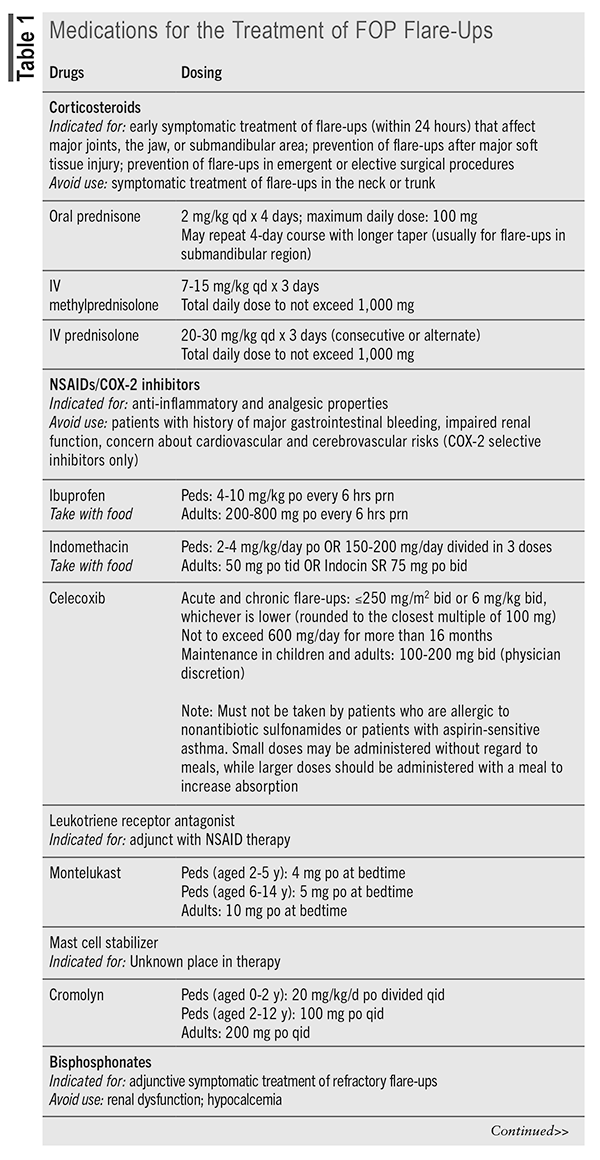
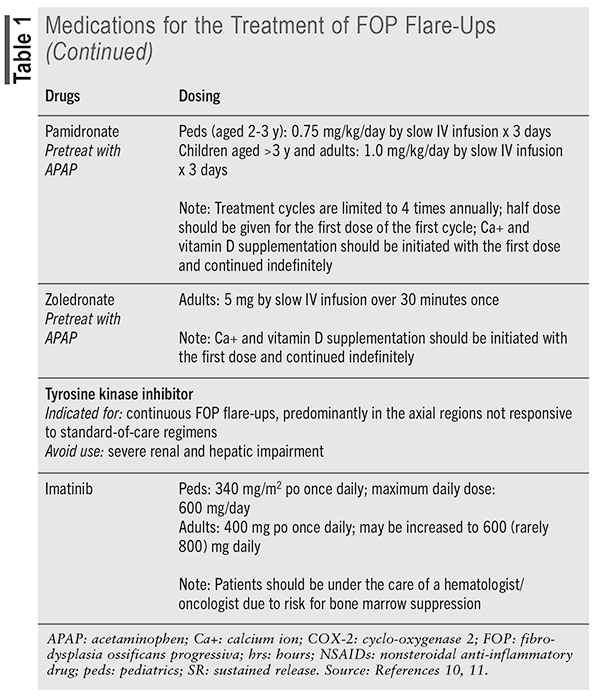
Novel Agent: Palovarotene (Sohonos) received FDA approval in October 2023 and became the first medication indicated for the treatment of FOP in females aged 8 years and older and males aged 10 years and older.13 It is a retinoic acid receptor gamma selective agonist that prevents the activation of chondrocytes by the erroneously activated BMP signaling cascade in patients with FOP. Palovarotene is contraindicated in pregnancy and breastfeeding and should not be used in patients with a history of allergy to retinoids.14
Patients receive maintenance and flare-up treatment (TABLE 2), and dosage reductions are required for patients who experience adverse effects or who are concomitantly taking a moderate cytochrome P450, family 3, subfamily A inhibitor. The capsule should be taken with food at the same time each day and can be swallowed whole or opened and contents sprinkled on one teaspoon of soft food.
Mucocutaneous adverse reactions (e.g., dry skin/lip, pruritus, rash, alopecia, skin exfoliation, dry eye) were most commonly reported in clinical trials, as well as pain (myalgia, musculoskeletal), headache, peripheral edema, fatigue, and night vision loss. Patients should be cautioned to avoid excessive exposure to sun (photosensitivity) and to be aware of the increased risk of fractures and exacerbation of psychiatric disorders.
CHAPLE Disease
Disease Overview: CHAPLE (complement hyperactivation, angiopathic thrombosis and protein-losing enteropathy) is a rare genetic disease marked by CD55 deficiency.15,16 Patients who are affected by the disease are homozygous carriers of loss-of-function CD55 variants, which has only been found in those of Moroccan, Syrian, or Turkish ancestry. CD55 is one of three regulatory proteins that inhibit unwanted complement activation on host/self cells.17 Without regulation, complements produce bioactive peptides, or anaphylatoxins, that alter innate and adaptive immune responses. This leads to a membrane attack complex (MAC) that causes cell and pathogen lysis. Loss of CD55 function is associated with complement and innate immunity damage to intestinal lymph vessels, resulting in protein-losing enteropathy.
Patients with CHAPLE disease present with hypoproteinemia, edema, malnutrition, hypogammaglobulinemia, and gastrointestinal symptoms (abdominal pain, vomiting, and diarrhea) at an early age (infancy to early childhood).15,17 Micronutrient deficiencies of iron, ferritin, calcium, magnesium, folate, vitamin D, and vitamin B12 are caused by chronic malabsorption, as are anemia and growth retardation. An increased risk of infection has been noted and attributed to the hypogammaglobulinemia. Thrombophilia has been shown to cause severe thrombotic vascular occlusions that were unresponsive to surgery and anticoagulation; as such, early death related to severe thrombotic events is presumed to be part of disease progression.
General Management: Prior to gaining a better understanding of the pathophysiology underlying CHAPLE disease, patients were broadly managed with supportive care.15,18 Albumin infusions are utilized to combat the hypoproteinemia that is due to protein-losing enteropathy. Immunosuppressive medications target the complement and innate immunity–mediated damage and inflammation. IV immune globulin transfusions decrease the frequency and severity of infections. To combat malabsorption, patients are given vitamin and micronutrient supplementation and should consume a protein-rich diet with medium-chain triglycerides. In those with severe anemia that is unresponsive to vitamin and mineral supplementation, blood transfusions are required. Lastly, surgical resection of lymphangiectatic intestinal segments may help with symptom resolution but is typically done only in the presence of a bowel obstruction.
Novel Agent: Pozelimab-bbfg (Veopoz) is a human monoclonal immunoglobulin G4P antibody that blocks the activation of complement component C5, thereby preventing MAC formation.18,19 In August 2023, it became the first FDA-approved medication for the treatment of CHAPLE disease. Previous off-label studies looked at the use of eculizumab, also a C5 complement inhibitor, but dosing challenges proved to be an issue. An open-label phase II and phase III study of pozelimab-bbfg performed in 10 patients showed normalization of albumin levels, improvement or no worsening in clinical symptoms (bowel movements, facial edema, peripheral edema), catch-up growth, and decreased concentrations of complement markers. Pozelimab requires an IV infusion and SC weekly injections and may then be used in patients aged 1 year and older (TABLE 2). Patients should be vaccinated against meningococcal infections prior to drug initiation or as soon as possible if urgent therapy is required. The most common adverse effects in the trial were upper respiratory tract infection, fracture, urticaria, and alopecia.
Primary Hyperoxaluria Type 1
Disease Overview: Primary hyperoxaluria (PH) is a group of rare diseases that are characterized by an overproduction of endogenous oxalate due to impaired hepatic glyoxylate metabolism.20,21 The PH (1-3) group of autosomal recessive disorders is differentiated by the location of the genetic mutation; PH1 is the result of a mutation in the AGXT gene that codes for liver-specific alanine: glyoxylate aminotransferase (AGT). The result is an increase in endogenous glyoxylate, which undergoes hepatic oxidation to oxalate for renal elimination. PH1 is the most frequent (about 80% of cases) and the most devastating form of the disease.
Overproduction of oxalate renders patients at risk for precipitation with calcium (CaOx) in the tubular lumen and interstitial tissue of the kidneys.20-22 The resulting recurrent nephrolithiasis and/or nephrocalcinosis induces severe pain, hematuria, and inflammation, predisposing patients to a progressive decline in renal function and failure. Without a means to eliminate oxalate, plasma levels rise, placing the patients at risk for CaOx deposits in various tissues (bone, retina, myocardium, vessel walls, skin). The age at which stone development occurs differs for each patient; however, PH should be considered in any child presenting with a kidney stone and adults with recurrent stones. Urine and plasma oxalate levels, combined with elevated glycolate, will be seen in patients with PH1, with genetic testing making the confirmatory diagnosis.
General Management: As with many other rare diseases, supportive treatment was the only treatment available.20,23 For patients with PH1, hyperhydration, use of crystallization inhibitors, dialysis, and transplant were the mainstays of treatment. Fluid intakes of 3.5 to 4 liters per day for adults and 1.5 to 3 L/m2 body surface area for children are recommended, with a goal urine output of 2.5 L/hour to enhance urine dilution of oxalate. Urinary alkalizers, such as potassium citrate, potassium bicarbonate, and sodium citrate, may bind excess calcium and reduce formation of CaOx; therefore, a trial is recommended for any patient who is suspected to have PH1. Similarly, pyridoxine supplementation should be trialed at a dose of 5 mg/kg for those suspected of having PH1. Pyridoxine is a cofactor for AGT, and supplementation may increase the activity of mistargeted AGT and decrease oxalate production in some variants of AGXT. Response to pyridoxine therapy is defined as a >30% relative decrease in urine oxalate from baseline within 6 months. Dialysis may be used in patients with preserved renal function to aid with oxalate elimination and is required in patients with end-stage renal disease (ESRD). Liver transplantation is the only cure for PH1 and may be combined with a kidney transplant in patients with ESRD.
Novel Agent: Nedosiran (Rivfloza) is a double-stranded, small-interfering RNA (siRNA) that targets the hepatic lactate dehydrogenase A pathway, thereby decreasing oxalate production by preventing the conversion of glyoxylate.20,23,24 Lumasiran was the first siRNA approved for the treatment of PH1, and it works further upstream by inhibiting glycolate oxidase, the enzyme needed to convert glycolate into glyoxylate. Both agents have been shown to decrease urine oxalate concentrations (primary efficacy outcome), with limited data suggesting a reduction in plasma oxalate concentrations for patients with impaired renal function. Long-term data are still being collected to determine the effect of these agents on stone recurrence, regression of nephrocalcinosis, and maintenance of kidney function. Nedosiran is a once-monthly SC injection approved for patients aged 9 years and older with PH1 (TABLE 2). Injection-site reactions, erythema, pain, bruising, and rash were the only adverse effects seen in clinical trials, and these reactions did not lead to treatment discontinuation.
Duchenne Muscular Dystrophy
Disease Overview: Duchenne muscular dystrophy (DMD) is an X-linked recessive neuromuscular disorder that has a reported prevalence of 15.9 cases per 100,000 live male births in the U.S.25,26 It is the result of a genetic mutation that causes an absence or deficiency of dystrophin, a cytoskeletal protein responsible for the strength, stability, and functionality of myofibers. This deficiency, coupled with a series of damaging processes, is responsible for the progressive weakening and degeneration seen in patients with DMD. Although female carriers are largely unaffected, approximately 10% may show disease manifestations that include cognitive and/or cardiac function.27
Motor delays, often most noticeable at age 5 years, are the predominate first symptom that spurs the diagnosis in males.27 The clinical course of muscle decline is variable for all; left untreated, loss of ambulation necessitating the use of a wheelchair occurs before the teenage years. Subsequent respiratory, orthopedic, and cardiac complications emerge, contributing to early mortality at around age 19 years. As more has been understood about DMD and its progression, disease management has helped to increase the lifespan of afflicted patients, who are now living into their 30s.
General Management: Glucocorticoids and physiotherapy remain the mainstays of early DMD treatment.25 Prednisone (0.3-1.5 mg/kg/day) and deflazacort (0.9-1 mg/kg/day) have been associated with improved strength, age at loss of ambulation, timed motor function, reduced risk of scoliosis surgery, improved pulmonary function, and delayed onset of cardiomegaly.28
In October 2023, the FDA approved a novel corticosteroid, vamorolone (Agamree), for the treatment of DMD in patients aged 2 years and older dosed at 6 mg/kg to a maximum of 300 mg.29 The downside to the long-term use of glucocorticoids includes weight gain, hirsutism, cushingoid appearance, cataracts, skin fragility, and osteoporosis. Through diet and supplements, patients should maintain a total calcium intake of 1,200 mg/day and vitamin D intake of 800 IU/day.
Given the nature of DMD progression, multidisciplinary teams will include endocrine, gastrointestinal (GI) and nutritional, respiratory, cardiac, and orthopedic management.25 While this team is initially brought onboard to assist with organ system monitoring, progressive muscular decline will often lead to the need for tube feeding, ventilatory support, orthopedic support devices, or surgery. Patients should be initiated on an angiotensin-converting enzyme inhibitor or angiotensin blocker by age 10 years and monitored/treated for heart failure. Psychosocial support should be given to the patient and caregivers, as the burden of disease is large, particularly once ambulation is lost.
Novel Agent: Givinostat (Duvyzat) is a histone deacetylase inhibitor that is approved for patients aged 6 years and older with DMD.30 Although the mechanism through which it exerts an effect in DMD is unknown, when given in addition to glucocorticoids, the decline in 18-month muscle function from baseline was significantly decreased (evaluated by a four-stair climb). Serious adverse effects include thrombocytopenia, elevated triglycerides, GI disturbances (diarrhea, nausea/vomiting, abdominal pain), and QTc prolongation. Additional adverse reactions include muscle pain, pyrexia, and fatigue. Givinostat is an oral suspension (8.86 mg/mL) that is dosed based on actual body weight and administered twice daily with food (TABLE 2). Prior to treatment initiation, baseline platelet count and triglycerides should be obtained, as well as an ECG in patients with history of underlying cardiac disease or who are on other QTc-prolonging medications. Dose adjustments based on adverse effects are provided by the manufacturer.
Conclusion
Through scientific advances within the past century, the landscape of rare disease care is rapidly changing. As new, rarely utilized medications with complex mechanisms of action become available, pharmacists are needed to assist with patient and provider education and ensure safety.
REFERENCES
1. National Center for Advancing Translational Sciences. About GARD. https://rarediseases.info.nih.gov/about. Accessed May 29, 2024.
2. Aronson J. Rare diseases, orphan drugs, and orphan diseases. BMJ. 2006;333(7559):127.3. Swann J. The story behind the Orphan Drug Act. www.fda.gov/industry/fdas-rare-disease-day/story-behind-orphan-drug-act. Accessed May 29, 2024.4. Fermaglich LJ, Miller KL. A comprehensive study of the rare diseases and conditions targeted by orphan drug designations and approvals over the forty years of the Orphan Drug Act. Orphanet J Rare Dis. 2023;18(1):163.
5. Barrett JS, Betourne A, Walls RL, et al. The future of rare disease drug development: the rare disease cures accelerator data analytics platform (RDCA-DAP). J Pharmacokinet Pharmacodyn. 2023;50(6):507-519.
6. de Ruiter RD, Smilde BJ, Pals G, et al. Fibrodysplasia ossificans progressiva: what have we achieved and where are we now? Follow-up to the 2015 Lorentz Workshop. Front Endocrinol (Lausanne). 2021;12:732728.
7. Kaliya-Perumal AK, Carney TJ, Ingham PW. Fibrodysplasia ossificans progressiva: current concepts from bench to bedside. Dis Model Mech. 2020;13(9):dmm046441.
8. Smilde B, Botman E, de Ruiter R, et al. Monitoring and management of fibrodysplasia ossificans progressiva: current perspectives. Orthop Res Rev. 2022;14:113-120.
9. Pignolo RJ, Shore EM, Kaplan FS. Fibrodysplasia ossificans progressiva: clinical and genetic aspects. Orphanet J Rare Dis. 2011;6(1):80.
10. Kannu P, Levy CE. Evolving management of fibrodysplasia ossificans progressiva. J Pediatr. 2021;232:S9-S15.
11. Kaplan FS, Al Mukaddam M, Baujat G, et al. The medical management of fibrodysplasia ossificans progressive: current treatment considerations. Proc Intl Clin Council FOP. 2022;2:1-127.
12. International Clinical Council (ICC) on FOP. Statement regarding off label medications for the management of FOP, from the International Clinical Council (ICC) on FOP. www.iccfop.org/dvlp/wp-content/uploads/2024/03/20240319-ICC-Off-Label-Statement-FINAL.pdf. Accessed May 30, 2024.
13. Ipsen Biopharmaceuticals Canada Inc. Product Monograph: SOHONOS palovarotene capsules. November 17, 2023. www.ipsen.com/websites/Ipsen_Online/wp-content/uploads/sites/61/2024/01/05103949/PM-Sohonos-EN-17Nov2023.pdf. Accessed May 30, 2024.
14. SOHONOS (palovarotene) product information. Cambridge, MA: Ipsen Biopharmaceuticals, Inc. August 2023. www.accessdata.fda.gov/drugsatfda_docs/label/2023/215559s000lbl.pdf. Accessed May 31, 2024.
15. Ozen A, Comrie WA, Ardy RC, et al. CD55 deficiency, early-onset protein-losing enteropathy, and thrombosis. N Engl J Med. 2017;377(1):52-61.
16. Belot A, Benezech S, Tusseau M. A new drug for rare diseases: pozelimab for CHAPLE disease. Lancet. 2024;403(10427):592-593.
17. Ozen A, Kasap N, Vujkovic-Cvijin I, et al. Broadly effective metabolic and immune recovery with C5 inhibition in CHAPLE disease. Nat Immunol. 2021;22(2):128-139.
18. Ozen A, Chongsrisawat V, Sefer AP, et al. Evaluating the efficacy and safety of pozelimab in patients with CD55 deficiency with hyperactivation of complement, angiopathic thrombosis, and protein-losing enteropathy disease: an open-label phase 2 and 3 study. Lancet. 2024;403(10427):645-656.
19. VEOPOZ (pozelimab-bbfg) product information. Basking Ridge, NJ: Regeneron Pharmaceuticals, Inc. August 2023. www.accessdata.fda.gov/drugsatfda_docs/label/2023/761339s000lbl.pdf. Accessed June 1, 2024.
20. Groothoff JW, Metry E, Deesker L, et al. Clinical practice recommendations for primary hyperoxaluria: an expert consensus statement from ERKNet and OxalEurope. Nat Rev Nephrol. 2023;19(3):194-211.
21. Hoppe B, Martin-Higueras C. Improving treatment options for primary hyperoxaluria. Drugs. 2022;82(10):1077-1094.
22. Cochat P, Hulton SA, Acquaviva C, et al. Primary hyperoxaluria type 1: indications for screening and guidance for diagnosis and treatment. Nephrology Dialysis Transplantation. 2012;27(5):1729-1736.
23. Gupta A, Somers MJG, Baum MA. Treatment of primary hyperoxaluria type 1. Clin Kidney J. 2022;15(Suppl 1):i9-i13.
24. Rivfloza (nedosiran) product information. Costa Mesa, CA: Pyramid Laboratories. September 2023. www.accessdata.fda.gov/drugsatfda_docs/label/2023/215842s000lbl.pdf. Accessed June 1, 2024.
25. Birnkrant DJ, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018;17(3):251-267.
The content contained in this article is for informational purposes only. The content is not intended to be a substitute for professional advice. Reliance on any information provided in this article is solely at your own risk.
To comment on this article, contact rdavidson@uspharmacist.com.



